CANNABINOIDS AND ANIMAL PHYSIOLOGY
2.1
Chapter 2.
Cannabinoids and Animal Physiology
Introduction
Much has been learned since the publication of the 1982 IOM
report on Marijuana and Health.a Although it was clear then that
most of the effects of marijuana were due to its actions on the
brain, there was little information about how THC acted on brain
cells (neurons), which cells were affected by THC, or even what
general areas of the brain were most affected by THC. Too little
was known about cannabinoid physiology to offer any scientific
insights into the harmful or therapeutic effects of marijuana.
That is no longer true. During the last 16 years, there have been
major advances in what basic science discloses about the potential
medical benefits of cannabinoids, the group of compounds related
to THC. Many variants are found in the marijuana plant, and other
cannabinoids not found in the plant have been chemically synthesized.
Sixteen years ago, it was still a matter of debate as to whether
THC acted nonspecifically by affecting the fluidity of cell membranes
or whether a specific pathway of action was mediated by a receptor
that responded selectively to THC (table 2.1).
Basic science is the wellspring for developing new medications
and is particularly important for understanding a drug that has
as many effects as marijuana. Even committed advocates of the
medical use of marijuana do not claim that all the effects of
marijuana are desirable for every medical use. But they do claim
that the combination of specific effects of marijuana enhances
its medical value. An understanding of those specific effects
is what basic science can provide. The multiple effects of marijuana
can be singled out and studied with the goals of evaluating the
medical value of marijuana and cannabinoids in specific medical
conditions, as well as minimizing unwanted side effects. An understanding
of the basic mechanisms through which cannabinoids affect physiology
permits more strategic development of new drugs and designs for
clinical trials that are most likely to yield conclusive results.
Research on cannabinoid biology offers new insights into clinical
use, especially given the scarcity of clinical studies that adequately
evaluate the medical value of marijuana. For example, despite
the scarcity of substantive clinical data, basic science has made
it clear that cannabinoids can affect pain transmission and specifically
that, cannabinoids interact with the brain's endogenous opioid
system, an important system for the medical treatment of pain
(see chapter 4).
a The field of neuroscience has grown substantially since the
publication of the 1982 IOM report. The number of members in the
Society for Neuroscience provide a rough measure of the growth
in research and knowledge about the brain: as of the middle of
1998, there are over 27,000 members, more than triple the number
in 1982.
2.2
The cellular machinery that underlies the response of the body
and brain to cannabinoids involves an intricate interplay of different
systems. This chapter reviews the components of that machinery
with enough detail to permit the reader to compare what is known
about basic biology with specific indications proposed for marijuana.
For some readers' that will be too much detail. Those readers
who do not wish to read the entire chapter should, nonetheless,
be mindful of the following key points in this chapter
· The most far-reaching of the recent advances in cannabinoid
biology are the identification of two types of cannabinoid receptors
(CB1 and CB2) and of anandamide, a substance naturally produced by the body that acts at the cannabinoid receptor, and has effects
similar to those of THC. The CB1 receptor is found primarily in
the brain, and mediates the psychological effects of THC. The
CB2 receptor is associated with the immune system, its role remains unclear.
· The physiological roles of the brain cannabinoid system in
humans are the subject of much active research, and not fully
known; however, cannabinoids likely have a natural role in pain
modulation, control of movement, and memory.
· Animal research has shown that the potential for cannabinoid
dependence exists, and cannabinoid withdrawal symptoms can be
observed. However, both appear to be mild compared to dependence
and withdrawal seen with other drugs.
· Basic research in cannabinoid biology has revealed a variety
of cellular pathways through which potentially therapeutic drugs
could act on the cannabinoid system. In addition to the known
cannabinoids, such drugs might include chemical derivatives of
plant-derived cannabinoids or of endogenous cannabinoids such
as anandamide, but would also include non-cannabinoid drugs that
act on the cannabinoid system.
This chapter summarizes the basics of cannabinoid biology -
as known today. It thus provides a scientific basis for interpreting
claims founded on anecdotes and for evaluating the clinical studies
of marijuana presented in chapter 4.
2.3
Table 2.1 Landmark Discoveries Since the 1982 IOM Report
Since the Previous IOM Report on Marijuana in 1982:
A Decade of Landmark Discoveries |
| Year |
Discovery |
Primary Investigators |
| 1986 |
Potent cannabinoid agonists are developed the key to discovering
the receptor |
M.R. Johnson and L.S. Melvin75 |
| 1988 | First conclusive evidence of specific cannabinoid receptors | A. Howlett and W. Devane36 |
| 1990 | The cannabinoid brain receptor (CB1) is cloned, its DNA sequence is identified, and its location in the brain is determined |
L. Matsuda et al,107 and M. Herkenham et al60 |
| 1992 | Anandamide is discovered - a naturally occurring substance in the brain that acts on cannabinoid receptors |
R. Mechoulam and W. Devane37 |
| 1993 | A cannabinoid receptor is discovered outside the brain; this receptor (CB2) is related to the brain receptor but is distinct |
S. Munro112 |
| 1994 | The first specific cannabinoid antagonist, SR141716A, is developed | M.Rinaldi-Carmona132 |
| 1998 | The first cannabinoid antagonist, SR144528, that can distinguish between CB1 and CB2 receptors discovered.
| M. Rinaldi-Carmona133 |
2.4
The Value of Animal Studies
Much of the research into the effects of cannabinoids on the
brain is based on animal studies. Many speakers in the public
workshops associated with this study argued that animal studies
of marijuana are not relevant to humans. While animal studies
are no substitute for clinical trials, they are a necessary complement.
Ultimately, every biologically active substance exerts its effects
at the cellular and molecular level, and at this level, the evidence
has shown remarkable consistency among mammals, even those as
different in body and mind as rats and humans. Animal studies
typically provide information about how drugs work that would
not be obtainable in clinical studies. At the same time, animal
studies can never completely inform us about the full range of
psychological and physiological effects of marijuana or cannabinoids
on humans.
The Active Constituents of Marijuana
 9-THC and 8-THC are the only compounds in the marijuana plant
that show all the psychoactive effects of marijuana. Because 9-THC
is much more abundant than 8-THC, the psychoactivity of marijuana
has been largely attributed to the effects of 9-THC 11-OH-9-THC
is the primary product of 9-THC metabolism by the liver and is
about three times as potent as 9-THC.128
9-THC and 8-THC are the only compounds in the marijuana plant
that show all the psychoactive effects of marijuana. Because 9-THC
is much more abundant than 8-THC, the psychoactivity of marijuana
has been largely attributed to the effects of 9-THC 11-OH-9-THC
is the primary product of 9-THC metabolism by the liver and is
about three times as potent as 9-THC.128
There have been considerably fewer experiments with cannabinoids
other than 9-THC although a few studies have been done to examine
whether other cannabinoids modulate the effects of THC or mediate
the non-psychological effects of marijuana. Cannabidiol (CBD)
does not have the same psychoactivity as THC, but it was initially
reported to attenuate the psychological response to THC in humans
81, 177 however, later studies reported that CBD did not attenuate
the psychological effects of THC.11, 69 One double-blind study
of eight volunteers reported that CBD can block the anxiety induced
by high doses of THC (0.5 mg/kg).177 There are numerous anecdotal
reports claiming that marijuana with relatively higher ratios
of THC:CBD is less likely to induce anxiety in the user than marijuana
with low THC:CBD ratios; but, taken together, the results published
thus far are inconclusive.
The most significant effects of cannabidiol (CBD) seem to be
its interference with drug metabolism in the liver, including
9-THC metabolism.14, 114 CBD exerts this effect by inactivating
cytochrome P450s, which are the most important class of enzymes
that metabolize drugs. Like many P450 inactivators, CBD can also
induce P450s after repeated doses.13 Experiments in which mice
were treated with CBD followed by THC showed that CBD treatment
was associated with a significant
2.5
increase in brain levels of THC and its major metabolites,
most likely because of its effects on decreasing the clearance
rate of THC from the body 15
In mice, THC inhibits the release of luteinizing hormone (LH),
the pituitary hormone that triggers the release of testosterone
from the testis in males, this effect is increased when THC is
given together with cannabinol or CBD.113
Cannabinol is considerably less active than THC in the brain,
but studies of immune cells have shown that it can modulate immune
function (see section on Cannabinoids and the Immune System).
In mice, cannabinol lowers body temperature and increases sleep
duration.175
The Pharmacological Toolbox
A researcher needs certain key tools in order to understand
how a drug acts on the brain. To appreciate the importance of
these tools, one must first understand some basic principles of
drug action. All recent studies have indicated that the behavioral
effects of THC are receptor-mediated.27 Neurons in the brain are
activated when a compound binds to its receptor, which is a protein
typically located on the cell surface. Thus, THC will exert its
effects only after binding to its receptor. In general, a given
receptor will accept only particular classes of compounds and
will be unaffected by other compounds.
Compounds that activate receptors are called agonists. Binding
to a receptor triggers an event or a series of events in the cell
that results in a change in the cell's activity, its gene regulation,
or the signals that it sends to neighboring cells (figure 2. 1).
This agonist-induced process is called signal transduction.
Another tool for drug research, which only recently became
available for cannabinoid research, are the receptor antagonists,
so-called because they selectively bind to a receptor that would
have otherwise been available for binding to some other compound
or drug. Antagonists block the effects of agonists and are tools
to identify receptor functions by showing what happens when a
receptor's normal functions are blocked. Agonists and antagonists
are both ligands; that is, they bind to receptors. Hormones, neurotransmitters,
and drugs can all act as ligands. Morphine and naloxone provide
a good example. A large dose of morphine acts as an agonist at
opioid receptors in the brain and interferes with, or even arrests,
breathing. Naloxone, a powerful opioid antagonist, blocks morphine's
effects on opiate receptors, thereby allowing an overdose victim
to resume breathing normally. Naloxone itself has no effect on
breathing.
Another key tool involves identifying the receptor protein
and determining how it works. That makes it possible to locate
where a drug activates its receptor in the brain -both the general
region of the brain and the cell type where the receptor is. The
way to find a receptor for a drug in the brain is to make the
receptor "visible" by attaching a radioactive or fluorescent
marker to the drug so that it can be detected.
2.6
Such markers show where in the brain it binds to the receptor,
but this is not necessarily the part of the brain where the drug
ultimately has its greatest effects.
Because drugs injected into animals must be dissolved in a
water-based solution, it is easier to deliver water-soluble molecules
than to deliver fat-soluble (lipophilic) molecules such as THC.
THC is so lipophilic that it can stick to glass and plastic syringes
used for injection. Because it is lipophilic, it readily enters
cell membranes and thus can cross the blood brain barrier easily.
(This barrier insulates the brain from many blood-borne substances.)
Early cannabinoid research was hindered by the lack of potent
cannabinoid ligands (THC binds to its cannabinoid receptors rather
weakly) and because they were not readily water-soluble. The synthetic
agonist, CP 55,940, which is more water-soluble than THC, became
the first useful research tool for studying cannabinoid receptors
because of its high potency, and the ability to label it with
a radioactive molecule, which enabled researchers to trace its
activity.
2.7
Figure 2.1 Diagram of neuron with synapse
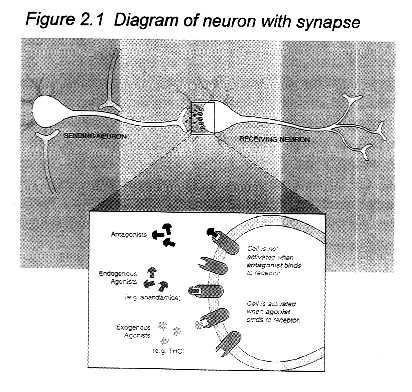
How receptors work: Individual nerve cells, or neurons, both
send and receive cellular signals to and from neighboring neurons,
but for the purposes of this diagram only one activity is indicated
for each cell. Neurotransmitter molecules (shown as black dots)
are released from the neuron terminal and move across the gap
between the "sending" and "receiving" neurons.
A signal is transmitted to the receiving neuron when the neurotransmitters
has bound to the receptor on its surface. The effects of a transmitted
signal include:
·Changing the cell's permeability to ions such as calcium and potassium.
·Turning a particular gene on or off.
·Sending a signal to another neuron.
·Increasing or decreasing the responsiveness of the cell to other
cellular signals.
Those effects can lead to cognitive, behavioral, or physiological
changes, depending on which neuronal system is activated.
The expanded view of the synapse illustrates a variety of ligands,
that is, molecules that bind to receptors. Anandamide is a substance
produced by the body that binds to and activates cannabinoid receptors;
it is an endogenous agonist. THC can also bind to and activate
cannabinoid receptors, but is not naturally found in the body;
it is an exogenous agonist. SR141617A binds to, but does not activate
cannabinoid receptors. In this way, it prevents agonists, such
as anandamide and THC, from activating cannabinoid receptors by
binding to the receptors without activating them; SR141617A is
an antagonist, but it is not normally produced in the body. Endogenous
antagonists, that is, those normally produced in the body, might
also exist, but none have been identified.
2.8
Cannabinoid Receptors
The cannabinoid receptor is a typical member of the largest
known family of receptors: the G-protein-coupled receptors with
their distinctive pattern in which the receptor molecule spans
the cell membrane seven times (figure 2.2). For excellent recent
reviews of cannabinoid receptor biology, see Childers and Breivogel,27 Abood and Martin,1 Felder and Glass,43 and Pertwee.124 Cannabinoid
receptor ligands bind reversibly (they bind to the receptor briefly
and then dissociate) and stereoselectively (when there are molecules
that are mirror images of each other, only one version activates
the receptor). Thus far, two cannabinoid receptor subtypes (CB1
and CB2) have been identified, of which only CB1 is found in the brain.
The cell responds in a variety of ways when a ligand binds
to the cannabinoid receptor (figure 2.3). The first step is activation
of G-proteins, the first components of the signal-transduction
pathway. That leads to changes in several intercellular components
- such as cyclic AMP and calcium and potassium ions - which ultimately
produce the changes in cell functions. The final result of cannabinoid
receptor stimulation depends on the particular type of cell,
the particular ligand, and the other molecules t hat might be
competing for receptor binding sites. Different agonists vary
in binding potency, which determines the effective dose of the
drug, and efficacy, which determines the maximal strength of the
signal that they transmit to the cell. The potency and efficacy
of THC are both relatively lower than those of some synthetic
cannabinoids; in fact, synthetic compounds are generally more
potent and efficacious than endogenous agonists.
CB1 receptors are extraordinarily abundant in the brain. They
are more abundant than most other G-protein-coupled receptors
and ten times more abundant than mu opioid receptors, the receptors
responsible for the effects of morphine.148
2.9
Figure 2.2 Cannabinoid receptors
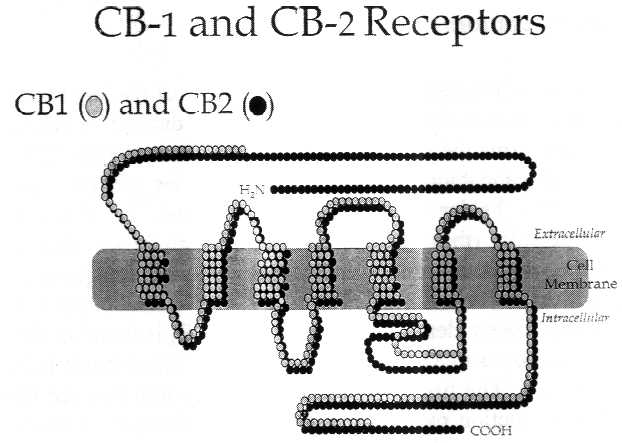
Receptors are proteins, and proteins are made up of strings
of amino acids. Each circle in the diagram represents one amino
acid. The shaded bar represents the cell membrane, which like
all cell membranes in animals, is largely composed of phospholipids.
Like many receptors, the cannabinoid receptors span the cell membrane;
some sections of the receptor protein are outside the cell membrane
(extrucellular), some are inside (intracellular). THC, anandamide,
and other known cannabinoid receptor agonists bind to the extracellular
portion of the receptor, thereby activating the signal pathway
inside the cell.
The CB1 molecule is larger than CB2. The receptor molecules
are most similar in four of the seven regions where they are embedded
in the cell membrane (known as the transmembrane regions). The
intracellular loops of the two cannabinoid receptor sub-types
are quite different, which might affect the cellular response
to the ligand, because these loops are known to mediate G-protein
signaling - that is, the next step in the cell signaling pathway
after the receptor. Receptor homology between the two receptor
sub-types is 44 percent for the full length protein, and 68 percent
within the seven transmembrane regions. The ligand binding sites
are typically defined by the extracellular loops and the transmembrane
regions.
2.10
Figure 2.3 How cannabinoids affected neuron signals
Figure legend. Intracellular events that happen when cannabinoid
agonists bind to receptors. Cannabinoid receptors are embedded
in the cell membrane where they are coupled to G-proteins (G)
and the enzyme, adenylyl cyclase (AC). Receptors are activated
when they bind with ligands such as anandamide or THC in this
case. This triggers a variety of reactions including inhibition
((-)) of AC which decreases the production of cAMP and cellular
activities dependent on cAMP, opening potassium (K+) channels
which decreases cell firing, and closing calcium (Ca2+) channels
which decreases the release of neurotransmitters. These changes
can influence cellular communication.
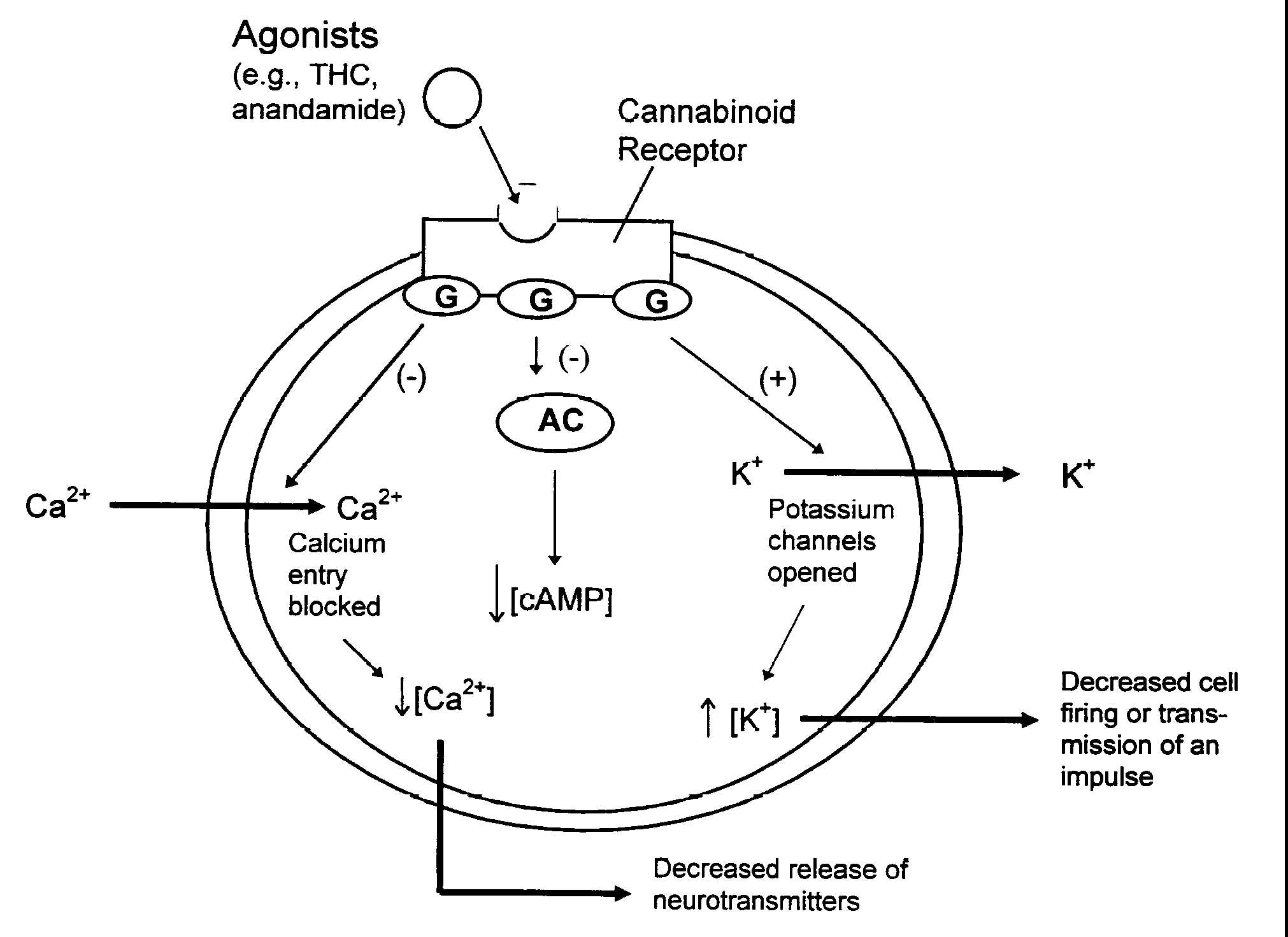 2.11
2.11
The cannabinoid receptor in the brain is a protein referred to
as CB1. The peripheral receptor (outside the nervous system),
CB2, is most abundant on cells of the immune system and is not
generally found in the brain.43, 124 Although no other receptor
subtypes have been identified, there is a genetic variant known
as CB1A (such variants are somewhat different proteins that have
been produced by the same genes via alternative processing). In
some cases, proteins produced via alternative splicing have different
effects on cells. It is not yet known whether there are any functional
differences between the two, but the structural differences raise
the possibility.
CB1 and CB2 are similar, but not as similar as members of many
other receptor families are to each other. On the basis of a comparison
of the sequence of amino acids that make up the receptor protein,
the similarity of the CB1 and CB2 receptors is 44 percent (figure
2.2). The differences between the two receptors indicate that
it should be possible to design therapeutic drugs that would act
only on one or the other receptor and thus would activate or attenuate
(block) the appropriate cannabinoid receptors. This offers a powerful
method for producing biologically selective effects. In spite
of the difference between the receptor subtypes, most cannabinoid
compounds bind with similar affinityb to both CB1 and CB2 receptors.
One exception is the plant-derived compound, cannabinol, which
shows greater binding affinity for CB2 than for CB1,112 although
another research group has failed to substantiate that observation.129
Other exceptions include the synthetic compound, WIN 55,212-2,
which shows greater affinity for CB2 than CB1, and the endogenous
ligands, anandamide and 2-AG, which show greater affinity for
CB1 than CB2.43 The search for compounds that bind to only one
or the other of the cannabinoid receptor types has been under
way for several years and has yielded a number of compounds that
are useful research tools and have potential for medical use.
Cannabinoid receptors have been studied most in vertebrates,
such as rats and mice. However, they are also found in invertebrates,
such as leeches and mollusks.156 The evolutionary history of vertebrates
and invertebrates diverged more than 500 million years ago, so
cannabinoid receptors appear to have been conserved throughout
evolution at least this long. This suggests that they serve an
important and basic function in animal physiology. In general,
cannabinoid receptor molecules are similar among different species.124
Thus, cannabinoid receptors likely fill many similar functions
in a broad range of animals, including humans.
b Affinity is a measure of how avidly a drug binds to a receptor.
The higher the affinity of a drug, the higher its potency; that
is, lower doses are needed to produce its effects.
2.12
The Endogenous Cannabinoid System
For any drug for which there is a receptor, the logical question
is, "Why does this receptor exist?" The short answer
is that there is probably an endogenous agonist (that is, a compound
that is naturally produced in the brain) that acts on that receptor.
The long answer begins with a search for such compounds in the
area of the body that produce the receptors and ends with a determination
of the natural function of those compounds. So far, the search
has yielded several endogenous compounds that bind selectively
to cannabinoid receptors. The best studied of them are anandamide37
and arachidonyl glycerol (2-AG).108 However, their physiological
roles are not yet known.
Initially, the search for an endogenous cannabinoid was based
on the premise that its chemical structure would be similar to
that of THC; that was reasonable, in that it was really a search
for another "key" that would fit into the cannabinoid
receptor "keyhole," thereby activating the cellular
message system. One of the intriguing discoveries in cannabinoid
biology was how chemically different THC and anandamide are. A
similar search for endogenous opioids (endorphins) also revealed
that their chemical structure is very different from the plant-derived
opioids, opium and morphine.
Further research has uncovered a variety of compounds with
quite different chemical structures that can activate cannabinoid
receptors (table 2.2 and figure 2.4) It is not yet known exactly
how anandamide and THC bind to cannabinoid receptors. Knowing
this should permit more precise design of drugs that selectively
activate the endogenous cannabinoid systems.
Anandamide
The first endogenous cannabinoid to be discovered was arachidonylethanolamine,
named anandamide from the Sanskrit word ananda, meaning "bliss."37
Compared with THC, anandamide has only moderate affinity for CB1,
and is rapidly metabolized by amidases (enzymes that remove amide
groups). Despite its short duration of action, anandamide shares
most of the pharmacological effects of THC37, 152 Rapid degradation
of active molecules is a feature of neurotransmitter systems that
allows them control of signal timing by regulating the abundance
of signaling molecules. It creates problems for interpreting the
results of many experiments and might explain why in vivo studies
with anandamide injected into the brain have yielded conflicting
results.
Anandamide appears to have both central (in the brain) and
peripheral (in the rest of the body) effects. The precise neuroanatomical
localization of anandamide and the enzymes that synthesize it
are not yet known. This information will provide
2.13
essential clues to the natural role of anandamide and an understanding
of the brain circuits in which it is a neurotransmitter. The importance
of knowing specific brain circuits that involve anandamide (and
other endogenous cannabinoid ligands) is that such circuits are
the pivotal elements for regulating specific brain functions,
such as mood, memory, and cognition. Anandamide has been found
in numerous regions of the human brain: hippocampus (and parahippocampic
cortex), striatum, and cerebellum; but it has not been precisely
identified with specific neuronal circuits. CB1 receptors are
abundant in these regions, and this further implies a physiological
role for endogenous cannabinoids in the brain functions controlled
by these areas. But, substantial concentrations of anandamide
are also found in the thalamus, an area of the brain that has
relatively few CB1 receptors.124
Anandamide has also been found outside the brain. It has been
found in spleen, which also has high concentrations of CB2 receptors;
and small amounts have been detected in heart.44
In general, the affinity of anandamide for cannabinoid receptors
is only one fourth to one-half that of THC (see table 2.3). The
differences depend on the cells or tissue that are tested and
on the experimental conditions, such as the binding assay used
(reviewed by Pertwee124).
The molecular structure of anandamide is relatively simple,
and it can be formed from arachidonic acid and ethanolamine. Arachidonic
acid is a common precursor of a group of biologically active
molecules known as eicosanoids, including prostaglandins.c Although
anandamide can be synthesized in a variety of ways, the physiologically
relevant pathway seems to be through enzymatic cleavage of N-arackidonyl-phosphatidyl-ethanolamine
(NAPE), which yields anandamide and phosphatidic acid (reviewed
by Childers and Breivogel27).
Anandamide can be inactivated in the brain via two mechanisms.
In one, anandamide is enzymatically cleaved to yield arachidonic
acid and ethanolamine- the reverse of what was initially proposed
as its primary mode of synthesis. In the other, it is inactivated
through neuronal uptake-i.e.., by being transported into the neuron,
which prevents its continuing activation of neighboring neurons.
c Eicosanoids all contain a chain of 20 carbon atoms, and are named after eikosi, the Greek word for twenty.
2.14
Table 2.2 Compounds that bind to cannabinoid receptors
| Compounds That Bind to Cannabinoid Receptorsd |
| Compound | Properties |
Agonists (Receptor activators) |
| Plant-derived compounds |
| 9-THC |
Main psychoactive cannabinoid in the marijuana plant; largely
responsible for psychological and physiological effects. (Except
in discussions of the different forms of THC, THC is used as a
synonym for 9-THC |
| 8-THC | Slightly less potent than 9-THC and much less abundant
in the marijuana plant, but otherwise similar. |
| 11-OH-9-THC |
Bioactive compound formed when the body breaks
down 9-THC. Presumed to be responsible for some of the effects of marijuana. |
| Cannabinoid agonists found in animals |
anandamide
(arachidonyl-
ethanolamide) |
Found in animals ranging from mollusks to mammals. Appears to be primary endogenous cannabinoid agonist in mammals. Chemical structure very different from plant cannabinoids, and related to prostaglandins. |
2-AG
(arachidonyl glycerol) | Endogenous agonist. Structurally similar to anandamide. More abundant but less potent than anandamide. |
| THC analogues |
| Dronabinol | Synthetic THC. Marketed in the US under the name Marinol® for nausea associated with chemotherapy and for AIDS-related wasting. |
| Nabilone | THC analogue. Marketed in the UK under the name Cesamet® for the same indications as dronabionol. |
| CP 55,940 | Synthetic cannabinoid; THC analogue; that is, it is structurally similar to THC |
| Levonantradol | THC analogue. |
| HU-210 | THC analogue, 100-800 fold greater potency than THC.97 |
| Chemical structure unlike THC or anandamide |
| WIN-55,212 | Chemical structure different from known cannabinoids, but binds to both cannabinoid receptors. Chemically related to
cyclo-oxygenase inhibitors, which include anti-inflammatory drugs. |
| Antagonists (Receptor Blockers) |
| SR 141716A | Synthetic CB1 antagonist, developed in 1994.132 |
| SR 144528 | Synthetic CB2 antagonist; developed in 1997.133 |
| d Sources: Mechoulam et al. 1998 109, Felder and Glass 199843;
BMA 199717 |
2.15
Figure 2.4 Chemical structures of compounds that bind to cannabinoid
receptors

Figure Legend. Selected cannabinoid agonists, or molecules
that bind to and activate cannabinoid receptors. THC is the primary
psychoactive molecule found in marijuana. CP 55,940 is a THC-analogue;
that is, its chemical structure is related to THC. Anandamide
and 2-arachidonyl glycerol (2-AG) are endogenous molecules, meaning
they are naturally produced in the body. Although the chemical
structure of WIN 55,212 is very different from either THC or anandamide,
it is also a cannabinoid agonists.
2.16
Table 2.3 Comparison of cannabinoid receptor agonists
Potency can be measured in a variety of ways, from behavioral
to physiological to cellular. This table shows potency in terms
of receptor binding, which is the most broadly applicable to the
many possible actions of cannabinoids. For example, anandamide
binds to the cannabinoid receptor only about half as avidly as
does THC. Measures of potency might include effects on activity
(behavioral) or hypothermia (physiological).
The apparently low potency of 2-AG may, however, be misleading.
A study published late in 1998, reports that 2-AG is found with
two other, closely related compounds that, by themselves, are
biologically inactive, but in the presence of those two compounds,
2-AG is only three times less active than THC.9 Further, 2-AG
is much more abundant than anandamide, although the biological
significance of this remains to be determined.
| Receptor Binding in Brain Tissue124 |
| Compound | Potency Relative to THC |
| CP 55,940 | 59 |
| 9-THC | 1 |
| Anandamide | 0.47 |
| 2-AG | 0.08 |
2.17
Other endogenous agonists
Several other endogenous compounds that are chemically related
to anandamide and that bind to cannabinoid receptors have been
discovered, one of which is 2-AG.108 2-AG is closely related to
anandamide and is even more abundant in the brain. At time of
this writing, all known endogenous cannabinoid receptor agonists
(including anandamide) are eicosanoids, which are arachidonic
acid metabolites. Arachidonic acid (a free fatty acid) is released
via hydrolysis of membrane phospholipids.
Other, non-eicosanoid, compounds that bind cannabinoid receptors
have recently been isolated from brain tissue, but they have not
been identified, and their biological effects are under investigation.
This is a fast-moving field of research, and no review over six
months old can be fully up-to-date.
The endogenous compounds that bind to cannabinoid receptors
probably perform a broad range of natural functions in the brain.
This neural signaling system is rich and complex, and has many
subtle variations' many of which await discovery. In the next
few years, much more will likely by known about these naturally
occurring cannabinoids.
Some effects of cannabinoid agonists are receptor-independent.
For example, both THC and CBD can be neuroprotective through their
antioxidative activity; that is, they can reduce the toxic forms
of oxygen that are released when cells are under stress.54 Other
likely examples of receptor-independent cannabinoid activity are
modulation of activation of membrane-bound enzymes (e.g., ATPase),
arachidonic acid release, and perturbation of membrane lipids.
An important caution in interpreting those reports is that concentrations
of THC or CBD used in cellular studies, such as these, are generally
much higher than the concentrations of THC or CBD in the body
that would likely be achieved by smoking marijuana.
Novel targets for therapeutic drugs
Drugs that alter the natural biology of anandamide, or other
endogenous cannabinoids, might have therapeutic uses (table 2.4).
For example, drugs that selectively inhibit neuronal uptake of
anandamide would increase the brain's own natural cannabinoids
and mimic some of the effects of THC. A number of important psychotherapeutic
drugs act by inhibiting neurotransmitter uptake. For example,
antidepressants like fluoxetine (Prozac®) inhibit serotonin
uptake, and are known as selective serotonin re-uptake inhibitors,
or SSRI's. Another way to alter levels of endogenous cannabinoids
would be to develop drugs that act on the enzymes involved in
anandamide synthesis. Some anti-hypertensive drugs work by inhibiting
2.18
enzymes involved in the synthesis of endogenous hypertensive agents.
Fore example, anti-converting enzyme (ACE) inhibitors are used
in hypertensive patients to interfere with the conversion of angiotensin
I, which is inactive, to the active hormone, angiotensin II
2.19
Table 2.4. Cellular processes that can be targeted for drug development
TABLE LEGEND. Endogenous cannabinoids are part of a cellular
signaling system. This table lists categories of natural processes
that regulate such systems, and shows the results of altering
those processes.
| Cellular Processes That Can Be Targeted for Drug Development |
| Drug action | . | Biological Result |
| Block synthesis |
Synthesis of bioactive compounds is a continuous process and is
one means by which concentrations of that compound are regulated. |
Weaker signal, due to decreased agonist concentration |
| Inhibit degradation | Chemical breakdown is one method the body uses to inactivate endogenous substances. |
Stronger signal, due to increased agonist concentration increased. |
| Facilitate neuronal uptake |
Neuronal uptake is one of the natural ways in which a receptor
agonist is inactivated. |
Stronger signal, due to increased amount of time during which
agonist is present in the synapse where it can stimulate the receptor |
2.20
Sites of Action
Cannabinoid receptors are particularly abundant in some areas
of the brain. The normal biology and behavior associated with
these brain areas are consistent with the behavioral effects produced
by cannabinoids (table 2.5 and figure 2.5). The highest receptor
density is found in cells of the basal ganglia that project locally
and to other brain regions. These cells include the substantia
nigra pars reticulata, entopeduncular nucleus, and globus pallidus,
regions that are generally involved in coordinating body movements.
Patients with Parkinson and Huntington disease tend to have impaired
functions in these regions.
CB1 receptors are also abundant in the putamen, part of the
relay system within the basal ganglia that regulates body movements,
the cerebellum, which coordinates body movements; the hippocampus,
which is involved in learning, memory, and response to stress;
and the cerebral cortex, which is concerned with the integration
of higher cognitive functions.
CB1 receptors are found on various parts of neurons, including
the axon, cell bodies, terminals, and dendrites.57, 165 Dendrites are generally the "receiving" part of a neuron, and
receptors on axons or cell bodies generally modulate other signals.
Axon terminals are the "sending" part of the neuron.
Cannabinoids like the inhibitory neurotransmitter  -aminobutyric
acid (GABA) -tend to inhibit neurotransmission, although the
results are somewhat variable. In some cases, cannabinoids diminish
the effects of the inhibitory neurotransmitter, -aminobutyric
acid (GABA),144 in other cases, cannabinoids can augment the effects
of GABA.120 The effect of activating a receptor depends on where
it is found on the neuron: if cannabinoid receptors are presynaptic
(on the "sending" side of the synapse) and inhibit the
release Of GABA, cannabinoids would diminish GABA effects; the
net effect would be stimulation. However, if cannabinoid receptors
are postsynaptic (on the "receiving" side of the synapse)
and on the same cell as GABA receptors, they will probably mimic
the effects of GABA; in that case, the net effect would be inhibition.120,
144, 160
-aminobutyric
acid (GABA) -tend to inhibit neurotransmission, although the
results are somewhat variable. In some cases, cannabinoids diminish
the effects of the inhibitory neurotransmitter, -aminobutyric
acid (GABA),144 in other cases, cannabinoids can augment the effects
of GABA.120 The effect of activating a receptor depends on where
it is found on the neuron: if cannabinoid receptors are presynaptic
(on the "sending" side of the synapse) and inhibit the
release Of GABA, cannabinoids would diminish GABA effects; the
net effect would be stimulation. However, if cannabinoid receptors
are postsynaptic (on the "receiving" side of the synapse)
and on the same cell as GABA receptors, they will probably mimic
the effects of GABA; in that case, the net effect would be inhibition.120,
144, 160
CB1 is the predominant brain cannabinoid receptor. CB2 receptors
have not generally been found in the brain, but there is one isolated
report suggesting some in mouse cerebellum.150 CB2 is found primarily
on cells of the immune system. CB1 receptors are also found in
immune cells, but CB2 is considerably more abundant there (table
2.6) (reviewed by Kaminski80 in 1998).
As can be appreciated in the next section, the presence of
cannabinoid systems in key brain regions is strongly tied to the
functions and pathology associated with those regions. The clinical
value of cannabinoid systems is best understood in the context
of the biology of these brain regions.
2.21
Table 2.5 Brain regions in which cannabinoid receptors are
abundante
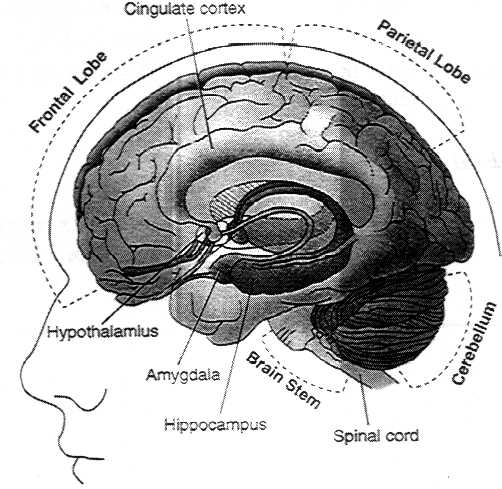
| Brain Region | Functions Associated with Region |
| Brain regions in which cannabinoid receptors are abundant |
Basal ganglia
Substantia nigra pars reticulate
Entopeduncular nucleus
Globus pallidus
Putamen |
Movement control |
| Cerebellum | Body-movement coordination |
| Hippocampus | Learning and memory, stress |
| Cerebral cortex, especially cingulate, frontal, and parietal regions | Higher cognitive functions |
| Nucleus accumbens | Reward center |
| Brain regions in which cannabinoid brain receptors are moderately concentrated |
| Hypothalamus | Body housekeeping functions (body-temperature regulation, salt and water balance, reproductive function) |
| Amygdala | Emotional response, fear |
| Spinal cord | Peripheral sensation, including pain |
| Brain Stem | Sleep and arousal, temperature regulation, motor control |
| Central gray | Analgesia |
| Nucleus of the solitary tract | Visceral sensation, nausea and vomiting |
e Based on reviews by Pertwee 1997 124 and Herkenham 199557 This table will be accompanied by a figure.
2.22
Figure 2.5. Location of brain regions in which cannabinoid
receptors are abundant.
See table 2.5 for summary of functions associated with those
regions.
 2.23
2.23
Table 2.6 Summary table of cannabinoid receptors
| . |
CB1 | CB2 |
| Effects of various cannabinoids |
| 9-THC | Agonist | Weak antagonist |
| Anandamide | Agonist | Agonist |
| Cannabinol (CBN) | Weak agonist |
Agonist; greater affinity for CB2 than for CB1 |
| Cannabidiol (CBD) | Does not bind to receptor | Does not bind to receptor |
| Receptor distribution |
| Areas of greatest abundance | Brain |
Immune system, especially B cells and natural killer cells |
2.24
Cannabinoid Receptors and Brain Functions
Motor effects
Marijuana affects psychomotor performance in humans. The effects
depend both on the nature of the task and the experience with
marijuana. In general, effects are clearest in steadiness (body
sway and hand steadiness) and in motor tasks that require attention.
The results of testing Cannabinoids in rodents are much clearer.
Cannabinoids clearly affect movement in rodents, but the effects
depend on the dose: low doses stimulate and higher doses inhibit
locomotion.111, 159 Cannabinoids mainly inhibit the transmission
of neural signals, and they inhibit movement through their actions
on the basal ganglia and cerebellum, where cannabinoid receptors
are particularly abundant (figure 2.6a and 2.6b). Cannabinoid
receptors are also found in the neurons that project from the
striatum and subthalamic nucleus, which inhibit and stimulate
movement, respectively.58, 101
Cannabinoids decrease both the inhibitory and stimulatory inputs
to the substantia nigra, and therefore might provide dual regulation
of movement at this nucleus. In the substantia nigra, Cannabinoids
decrease transmission from both the striatum and the subthalamic
nucleus.141 The globus pallidus has been implicated in mediating
the cataleptic effects of large doses of Cannabinoids in rats.126
(Catalepsy is a condition of diminished responsiveness usually
characterized by trancelike states and waxy rigidity of the muscles.)
Several other brain regions - the cortex, the cerebellum, and
the neural pathway from cortex to striatum - are also involved
in the control of movement and contain abundant cannabinoid receptors.52, 59, 101 They are, therefore, possible additional sites that might underlie the effects of Cannabinoids on movement.
2.25
Figure 2. 6a & b Diagrams showing motor regions of the
brain
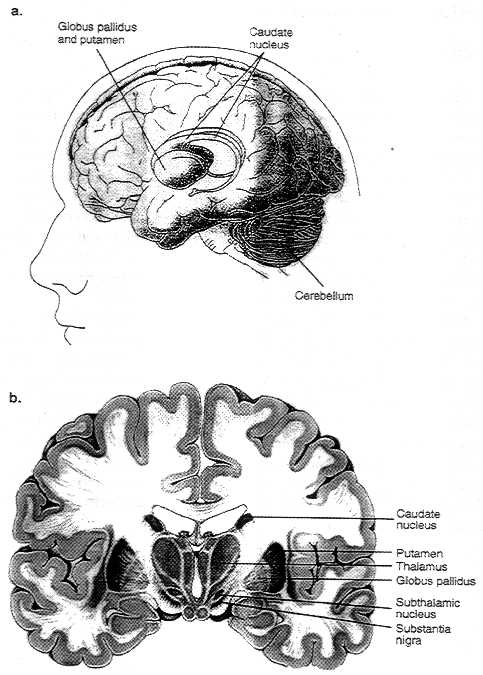
Figure 2.6. Basal ganglia are a group of three brain regions,
or nuclei - caudate, putamen, and globus pallidus. Figure 2.6a
is a 3-dimensional view showing the location of those nuclei in
the brain. Figure 2.6b shows those structures in a vertical cross-sectional
view The major output pathways of the basal ganglia arise from
the globus pallidus and pars reticula of the substantia nigra.
Their main target is the thalamus.
2.26
Memory effects
One of the primary effects of marijuana in humans is disruption
of short-term memory.68 That is consistent with the abundance
of CB1 receptors in the hippocampus, the brain region most closely
associated with memory. The effects of THC resemble a temporary
hippocampal lesion.63 Deadwyler and colleagues have demonstrated
that cannabinoids decrease neuronal activity in the hippocampus
and its inputs 23, 24, 83 In vitro, several cannabinoid ligands
and endogenous cannabinoids can block the cellular processes associated
with memory formation.29, 30, 116, 157, 163 Furthermore, cannabinoid agonists inhibit release of several neurotransmitters: acetylcholine from the hippocampus,49,50,51 norepinephrine from human and guinea pig (but not rat or mouse) hippocampal slices,143 and glutamate in cultured hippocampal cells.144 Cholinergic and noradrenergic
neurons project into the hippocampus, but circuits within the
hippocampus are glutamatergic.f Thus, cannabinoids could block transmission both into and within the hippocampus by blocking
presynaptic neurotransmitter release.
Pain
After nausea and vomiting, chronic pain was the condition cited
most often to the IOM study team as a medical use for marijuana.
Recent research presented below has shown intriguing parallels
with anecdotal reports of the modulating effects of cannabinoids
on pain - both the effects of cannabinoids acting alone and the
effects of their interaction with opioids.
Behavioral Studies
Cannabinoids reduce reactivity to acute painful stimuli in
laboratory animals. In rodents, cannabinoids reduced the responsiveness
to pain induced through various stimuli, including thermal, mechanical,
and chemical stimuli.12, 19, 46, 72, 96, 154, 174 Cannabinoids
were comparable with opiates in potency and efficacy in these
expeniments. 12, 72
Cannabinoids are also effective in rodent models of chronic
pain. Herzberg and coworkers found that cannabinoids can block
allodynia and hyperalgesia
fNeurons are often defined by the primary neurotransmitter released at their terminals. Thus, cholinergic neurons release acetylcholine, noradrenergic neurons release noradrenalin (also known as norepinephrine),
and glutamergic neurons release glutamate.
2.27
associated with neuropathic pain in rats.117 (Allodynia refers
to pain elicited by stimuli that are normally innocuous; hyperalgesia
refers to abnormally increased reactivity to pain.) This is an
important advance, because chronic pain frequently results in
a series of neural changes that increase suffering due to allodynia,
hyperalgesia, and spontaneous pain; furthermore' some chronic
pain syndromes are not amenable to therapy, even with the most
powerful narcotic analgesics.10
Pain perception is controlled mainly by neurotransmitter systems
within the central nervous system, and cannabinoids clearly play
a role in the control of pain in those systems.45 However, pain-relieving and pain-preventing mechanisms also occur in peripheral tissues,
and endogenous cannabinoids appear to play a role in peripheral
tissues. Thus, the different cannabinoid receptor subtypes might
act synergistically. Experiments in which pain is induced by injecting
dilute formalin into a mouse's paw have shown that anandamide
and palmitylethanolamide (PEA) can block peripheral pain.22, 73
22 Anandamide acts primarily at the CB1 receptor, whereas PEA has been proposed as a possible CB2 agonist, in short, there might be a biochemical basis for their independent effects. When injected
together, the analgesic effect is stronger than that of either
alone. That suggests an important strategy for the development
of a new class of analgesic drug: a mixture of CB1 and CB2 agonists. Because there are few, if any, CB2 receptors in the brain, it might be possible to develop drugs that enhance the peripheral analgesic effect while minimizing the psychological effects.
2.28
Neural sites of altered responsiveness to painful stimuli
The brain and spinal cord mediate cannabinoid analgesia. A
number of brain areas participate in cannabinoid analgesia and
support the role of descending pathways (neural pathways that
project from the brain to the spinal cord).103, 105 Although more
work is needed to produce a comprehensive map of the sites of
cannabinoid analgesia, it is clear that the effects are limited
to particular areas, most of which have an established role in
pain.
Specific sites where cannabinoids act to affect pain processing
include the periaqueductal gray,104 the rostral ventral medulla,
105, 110 and the thalamic nucleus submedius,102 the thalamic ventroposterolateral nucleus,102 dorsal horn of the spinal cord,64, 65 and peripheral sensory nerves.64, 65, 66, 131 Those nuclei also participate in opiate analgesia. Although similar to opiate analgesia, cannabinoid analgesia is not mediated by opioid receptors; morphine and cannabinoids sometimes act synergistically, and opioid antagonists generally have no effect on cannabinoid induced analgesia.171 However, a kappa-receptor antagonist has been shown to attenuate spinal, but not supraspinal, cannabinoid analgesia.153, 170, 171 (Kappa opioid receptors constitute one of the three major types of opioid receptors; the other two types are mu and delta receptors.)
2.29
Neurophysiology and neurochemistry of cannabinoid analgesia
Because of the marked effects of cannabinoids on motor function,
behavioral studies in animals alone cannot provide sufficient
grounds for the conclusion that cannabinoids depress pain perception.
Motor behavior is typically used to measure responses to pain,
but this behavior is itself affected by cannabinoids. Thus, experimental
results include an unmeasured combination of cannabinoid effects
on motor and pain systems. The effects on specific neural systems,
however, can be measured at the neurophysiological and neurochemical
levels. Cannabinoids decrease the response of immediate-early
genes (genes that are activated in the early or immediate stage
of response to a broad range of cellular stimuli) to noxious stimuli
in spinal cord, decrease response of pain neurons in the spinal
cord, and decrease the responsiveness of pain neurons in the ventral
posterolateral nucleus of the thalamus.67, 102 Those changes are
mediated by cannabinoid receptors, selective for pain neurons,
and unrelated to changes in skin temperature or depth of anesthesia,
and they follow the time course of the changes in behavioral responses
to painful stimuli, but not the time course of motor changes.67
Cannabinoids also modulate the responses of on-cells and off-cells
in the rostral ventral medulla in a manner that is very similar
to that of morphine.55, 110 These cells control pain transmission
at the level of the spinal cord.
Endogenous cannabinoids modulate pain
Endogenous cannabinoids can modulate pain sensitivity, through both central and peripheral mechanisms. For example, animal studies have shown that pain sensitivity can be increased when endogenous cannabinoids are blocked from acting at CB1 receptors 22, 62, 110, 130, 158 Administration of cannabinoid antagonists in either the spinal cord 130 or paw 22 increase the sensitivity of animals to pain. In addition, there is evidence that cannabinoids also act at the site of injury to reduce peripheral inflammation.131
Current data suggest the endogenous cannabinoid analgesic system might offer protection against the long-lasting central hyperalgesia and allodynia that sometimes follow skin or nerve injuries 130, 158 These results raise the possibility that therapeutic interventions
that alter the levels of endogenous cannabinoids might be useful
for managing pain in humans.
2.30
Chronic Effects of THC
Most substances of abuse produce tolerance, physical dependence, and withdrawal symptoms. Tolerance is the most common response to repetitive use of the same drug (not necessarily a drug of abuse) and is the condition in which, after repeated exposure to a drug, increasing doses are needed to achieve the same effect. Physical dependence develops as a result of tolerance (adaptation) produced by a resetting of homeostatic mechanisms in response to repeated drug use. It is important to reiterate that the phenomena of tolerance, dependence, and withdrawal are not associated uniquely with drugs of abuse. Many medications that are not addicting can produce these types of effects; examples of such medications include clonidine, propranolol, and tricyclic antidepressants. The following sections discuss what is known about the biological mechanisms that underlie on tolerance, reward, and dependence; clinical studies about those topics are discussed in chapter 3.
Tolerance
Chronic administration of cannabinoids to animals results in tolerance to many of the acute effects of THC, including memory disruption,34 decreased locomotion,2, 119 hypothermia,42, 125 neuroendocrine effects,134 and analgesia.4 Tolerance also develops to the cardiovascular and psychological effects of THC and marijuana in humans (see also discussion in chapter 3).55, 56, 76
Tolerance to cannabinoids appears to result from both pharmacokinetic
(how the drug is absorbed, distributed, metabolized, and excreted)
and pharmacodynamic (how the drug interacts with target cells)
changes. Chronic treatment with the cannabinoid agonist, CP 55,940,
increases the activity of the microsomal cytochrome P450 oxidative
system.31 Because this is the system through which drugs are metabolized
in the liver, this suggests pharrnacokinetic tolerance. Chronic
cannabinoid treatments also produce changes in brain cannabinoid
receptors and cannabinoid receptor mRNA levels, indicating that
pharmacodynamic effects are important, as well.
Most studies have found that brain cannabinoid receptor levels
usually decrease after prolonged exposure to agonists,42, 119, 136,
138 although some studies have reported increases 137 or no changes2
in receptor binding in brain. Differences among studies may be
due to the particular agonist tested, the assay used, brain region
examined, or treatment time. For example, the THC analogue, levonantradol,
produces a greater desensitization of adenylyl cyclase inhibition
than THC in cultured neuroblastoma cells,40 which may be explained
by the efficacy differences between these two agonists 18, 147
Furthermore, a time course study revealed differences in the rates
and magnitudes of receptor down-regulation across brain
2.31
regions.16 These findings suggest that tolerance to different
effects of cannabinoids develops at different rates
Chronic treatment with THC also produces variable effects on
cannabinoid-mediated signal transduction systems. Chronic THC
treatment produces significant desensitization of cannabinoid-activated
G-proteins in a number of rat brain regions.147 Moreover, the
time course of this desensitization varies across brain regions.16
It is difficult to extend the findings of these short-term
animal studies to human marijuana use In order to simulate long-term
use, the doses used in animal studies are higher than normally
achieved by smoking marijuana. For example, the average human
will feel "high" after a 0.06 mg/kg injection of THC,118
compared to 10-20 mg/kg/day used in many chronic studies in rats.
At the same time, doses of marijuana needed to observe behavioral
changes in rats (usually changes in locomotor behavior) are substantially
higher than doses at which people feel "high." In addition,
pharmacokinetics of THC distribution in the body are dramatically
different between rats and humans, as well as being highly dependent
on the THC delivery system - that is, whether it is inhaled, injected,
or swallowed. Nevertheless, it is likely that some of the same
biochemical adaptations to chronic cannabinoid administration
occur in both laboratory animals and humans, but the magnitude
of the effects in humans may be smaller in proportion to the respective
doses used.
Reward and dependence
Experimental animals that are given the opportunity to self-administer
cannabinoids generally do not choose to do so, which has led to
the conclusion that they are not reinforcing and rewarding.38
However, behavioral95 and brain stimulation94 studies have shown
that THC can be rewarding to animals. The behavioral study used
a "place-preference" test, in which an animal is given
repeated doses of a drug in one place, and is then given a choice
between a place where it did not receive the drug and one where
it did; the animals chose the place where they received the THC.
These rewarding effects are highly dose-dependent. In all models
studied, cannabinoids are only rewarding at mid-range; doses that
are too low are not rewarding, doses that are too high can be
aversive. Mice will self-administer the cannabinoid agonist, WIN
55,212, but only at low doses.106 This effect is specifically
mediated by CB1 receptors, and indicates that stimulation of those receptors is rewarding to the mice. Antagonism of cannabinoid
receptors is also rewarding in rats; in conditioned place-preference
tests, animals show a preference for the place they receive the
cannabinoid antagonist, SR141716A, at both low and high doses.140
Cannabinoids increase dopamine levels in the mesolimbic dopamine
system of rats, a pathway associated with reinforcement.25, 39,
161 However, the
2.32
mechanism by which THC increases dopamine levels appears to
be different from that of other abused drugs 51 g (see chapter 3 for further discussion of reinforcement).
Physical dependence on cannabinoids has only been observed
under experimental conditions of "precipitated withdrawal",
in which animals are first treated chronically with cannabinoids
and then given the CB1 antagonist, SR141716A.3, 166 The addition
of the antagonist accentuates any withdrawal effect by competing
with the agonist at receptor sites; that is, the antagonist helps
to clear agonists off and keep them off receptor sites. This suggests
that, under normal cannabis use, the long half-life and slow elimination
from the body of THC, and the residual bioactivity of its metabolite,
11-OH -THC, may prevent significant abstinence symptoms. The precipitated
withdrawal effects produced by SR141716A have some of the characteristics
of opiate withdrawal, but are not affected by opioid antagonists
and affect motor systems differently. An earlier study with monkeys
also suggested that abrupt cessation of chronic THC is associated
with withdrawal symptoms,8 Monkeys in that study were trained
to work for food after which they were given THC on a daily basis;
when the investigators stopped administering THC, the animals
stopped working for food.
A study in rats indicated that the behavioral cannabinoid withdrawal
syndrome correlates with stimulation of central amygdaloid corticotropin-releasing hormone release, consistent with the consequences of withdrawal
from other abused drugs.135 However, the withdrawal syndrome for
cannabinoids and the corresponding increase in corticotropin-releasing
hormone are only observed following administration of the CB1
antagonist, SR 141716A, to cannabinoidtolerant animals;3, 166
The implications of data based on precipitated withdrawal in animals
for human cannabinoid abuse have not been established.166 Furthermore, acute administration of THC also produces increases in corticotropin-releasing hormone and adrenocorticotropin release, both of which are stress-related hormones.71 This set of withdrawal studies may explain the generally aversive effects of cannabinoids in animals, and may indicate that the increase in corticotropin-releasing hormone is merely a rebound effect. Thus, while cannabinoids appear to be conforming to some of the neurobiological effects of other drugs abused by
humans, the underlying mechanisms of these actions and their significance
in determining the reinforcement and dependence liability of cannabinoids
in humans remain undetermined.
g These increases in dopamine are due to increases in the firing rate of dopamine cells in the ventral tegmental area by 9-THC47. However, these increases in firing rate in the ventral tegmental area could not be explained by increases in the firing of the A10 dopamine cell group, where other abused drugs have been shown
to act51.
2.33
Cannabinoids and the Immune System
The human body protects itself from invaders such as bacteria
and viruses through the elaborate and dynamic network of organs
and cells referred to as the immune system (see box on Cells of
the Immune System).
Cannabinoids, especially THC, can modulate the function of
immune cells in various ways - in some cases enhancing, and in
others diminishing the immune response 85 (summarized in table
2.7). However, the natural function of the cannabinoids in the
immune system is not known. Immune cells respond to cannabinoids
in a variety of ways, depending upon experimental factors such
as drug concentration, timing of drug delivery to leukocytes in
relation to antigen stimulation, and the type of cell function
analyzed. Although the chronic effects of cannabinoids on the
immune system have not been studied, based on acute exposure studies
in experimental animals it appears that the concentrations of
THC which modulate immunological responses are higher than those
required for psychoactivity.
2.34
Table 2.7 Effects of Cannabinoids on the Immune System
| Drug Tested |
Cell Types Tested or Type Drug of Animal Experiment |
Drug Concen- tration a |
Result | Reference |
THC
2-AG
11-OH-THC
CBN |
Lymphocytes and Splenocytes in vitro | 0.1-30 µM |
Higher doses suppress T cell proliferation |
Luo, 1992;
Pross,1992*
Klein, 1985%;
Specter,1990&
Lee, 1995*
Herring, 1998
|
THC
2-AG
Anandamide |
Lymphocytes and Splenocytes | 0.1-25 µM |
Lower doses increase T cell proliferation in vitro |
Luo,1992; Lee,1995* Pross,1992* |
| Splenocytes in vitro | 1-25 µM |
Little or no effect on T cell proliferation |
Lee,1995* Devane,1992 |
| THC, 11-OH-THC AG-2 |
Splenocytes in vitro | 3-30 µM |
Decrease B cell proliferation |
Klein,1985% Lee,1995* |
THC
CP 55,940
WIN 55,212-2 |
Lymphocytes in vitro |
0.1-100nM
[0.0001-0.1 µM] |
Increase B cell proliferation | Derocq, 1995 |
| THC |
Mice were injected with drug |
>5mg/kg |
Antibody production was suppressed | Baczynsky, 1983 Schatz,1993 |
| HU-210 | >0.05 mg/kg | Titishov,1989 |
THC
11-OH-THC
CBD
CP55,940
CBN |
Splenocytes in vitro | 1-30µM |
Antibody production was suppressed |
Klein,1990 Baczynasky,1983 Kaminski,1994 Kaminski,1992 Herring,1998 |
| THC | Rodents were injected with drug |
3mg/kg/day for 25days
40mg/kg/day for 2 days |
Repeated low doses or a high dose of THC suppress the activity
of natural killer cells |
Patel,1985 Klein,1987 |
| THC 1l-OH-THC | Natural killer cells in vitro |
0.1-32 µM | Doses of >=10 µM suppress natural killer cell cytolytic activity, doses <10 µM produced no effect | Klein,1987 Luo,1989 |
| THC | Peritoneal macrophages and monocytes |
3-30 µM |
Variable doses of THC suppress macophage functions in vitro | Lopez-Cepero, 1986
Specter,1991 Tang,1992 |
2.35
THC
CBD |
Mice injected with drug; in one case, in vitro tests done on spleens |
>5mg/kg for 4 days or 50 mg/kg every 5 days for up to 8 weeks |
THC suppresses normal immune response, interferons failed to increase when exposed to cytokine inducer, while CBD had no suppressive effect |
Cabral,1986 Blanchard,1986 |
THC
CBD |
Peripheral blood mononuclear cells in vitro |
<0.1 µM |
Increased interferon production | Warzl, 1991 |
| 30 µM |
Decreases interferon production | . |
THC
CBD |
Splenocytes and T cells in vitro | 10 µM |
Both THC and CBD suppress IL-2 secretion and the number of IL-2 transcripts |
Condie,1996 |
| THC |
Phorbol myristate acetate differentiated macrophage in vitro |
10-20 µM |
Increase in tumor necrosis factor production and IL-I supernatant bioactiviy |
Shivers, 1994 |
| THC |
Endotoxin-activated macrophages in vitro |
10-30 µM |
Increase processing and release of IL-I rather than cellular production of the IL-I | Zhu, 1994 |
| THC | Peritoneal macrophages in vitro |
10-30 µM | Increased IL-I bioactivity |
Klein, 1990 |
| THC |
Mice were injected with drug and either sublethal or lethal dose of Legionalla pneumophilia |
8mg/kg given before and after bacteria infection |
Cytokine-mediated septic shock and death occurs with exposure to sublethal dose of the bacteria | Klein, 1993 and 1994
Newton, 1994 |
| < 5 mg/kg doses. or one 8 mg/kg or 4 mg/kg dose given before bacteria infection | Survival occurs, but with greater susceptiblity to infection when challenged with bacteria and death when challenged with a lethal dose of bacteria |
| THC |
Immuno-deficient mice injected with drug and herpes simplex virus |
100mg/kg before and after virus infection |
Two high doses of THC potentiates the effects of herpes simplex and enhances the progression of death | Specter, 1991 |
| 100 mg/kg before virus infection | A single dose did not promote death |
* cell density dependent;
* mitogen dependent;
% % serum dependent;
& dependent on timing of drug exposure relative
to mitogen exposure.
a Drug concentrations are given in the standard format of molarity (M). A one molar solution is the molecular weight of the compound
(in grams) dissolved in 1 liter of water or other solvent. The
molecular weight of THC is 314 so a 1 molar solution would be
314 grams of THC dissolved in 1 liter of solution, a 10 µM solution
would be 3.14 mg THC/liter.
A 1-10 µM concentration will generally elicit a physiologically
relevant response in immune cell cultures. Higher doses are often
suspected of not being biologically meaningful, because they are
a much larger dose than would ever be achieved in the body. The
doses listed in this table are, for the most part, very high.
See text for further discussion.
2.36
Cells of the Immune System
The various organs of the immune system are positioned throughout
the body and include bone marrow, thymus, lymph nodes and spleen.
The cells of the immune system consist of white blood cells, or
leukocytes, which are formed in the bone marrow from stem cells
so-called because a great variety of cells descend from them (see
below). There are two kinds of leukocytes: Lymphocytes and phagocytes.
Lymphocytes consist of B cells, T cells,h and natural killer cells (NK), and the major phagocytes include monocytes, macrophages
and neutrophils. Phagocytes have many important roles in the immune
response, but most significantly they initiate these responses
by engulfing and digesting foreign substances (e.g., bacteria,
viruses, foreign proteins), or antigens, that enter the body.
Once digested, the antigen is exposed to specialized Iymphocytes
(i.e., B cells and T cells) so that antibodies and effector T
cells can be produced to help destroy any remaining antigens in
the body. Antibodies are proteins produced by B cells that bind
to antigens and promote antigen destruction. effector T cells
include killer T cells which attack and kill antigen laden cells,
and helper T cells, which secrete special proteins called cytokines
that promote antigen elimination. Natural killer cells are specialized
Iymphocytes that are also activated by antigen to either kill
infected targets or secrete immunoregulatory cytokines.
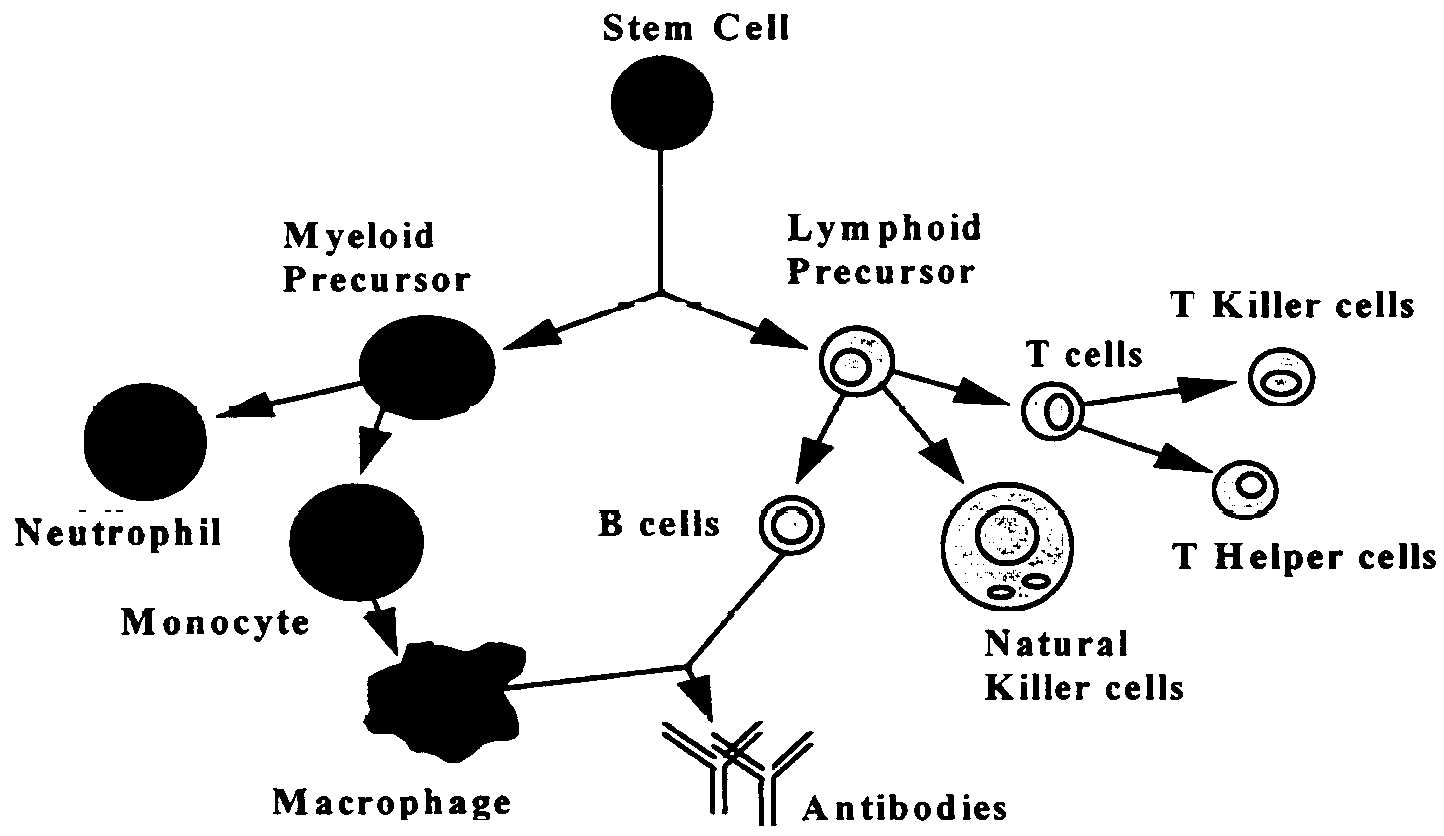
h The B and T refer to where the cells mature, either in the bone marrow (B) or thymus (T).
2.37
The CB2 receptor gene, which is not expressed in the brain,
is particularly abundant in immune tissues, with an expression
level 10-100 times higher than that of CB1 In spleen and tonsils,
the CB2 mRNAi content is equivalent to that of CB1 mRNA in the
brain.48 The rank order, from high to low, of CB2 mRNA levels
in immune cells is B-cells > natural killer cells >>
monocytes > polymorphonuclear neutrophil cells > T8 cells
in T4 cells. In tonsils, the CB2 receptors appear to be restricted
to B-lymphocyte-enriched areas. In contrast, CB1 receptors are
mainly expressed in the central nervous system and, to a lower
extent, in several peripheral tissues such as adrenal gland, heart,
lung, prostate, uterus, ovary, testis, bone marrow, thymus and
tonsils.
Cannabinoid Receptors and Intracellular Action in Immune Cells
CB2 appears to be the predominant gene expressed in resting
leukocytes.78, 112 The level of CB1 gene activity is normally
low in resting cells but increases with cell activation.32 Thus
the CB1 receptor might be important only when immune responses
are stimulated, but the physiological relevance of this observation
remains to be determined. Some of the cannabinoid effects observed
in immune systems, especially at high drug concentrations, are
likely mediated through non-receptor mechanisms, but these have
not yet been identified.4
Ligand binding to either the CB1 or CB2 receptors inhibits
adenylate cyclase, an enzyme that is responsible for cAMP production,
and is, thus, an integral aspect of intracellular signal transduction
(see figure 2.3).53, 79, 91, 122, 139, 151, 167 Increases in intracellular
cAMP concentrations lead to immune enhancement, while decreases
lead to an inhibition of immune responses.77 Cannabinoids inhibit
the rise in intracellular cAMP that normally results from leukocyte
activation, and this might be the pathway through which cannabinoids
suppress immune cell functions.28, 74, 167 In addition, cannabinoids
activate other molecular pathways such as the nuclear factor-kB
pathway and therefore these signals might be modified in drug
treated immune cells.33, 74
i After a gene is transcribed it is often spliced and modified
into mRNA, or message RNA. The CB-2 mRNA is the gene "message"
that moves from the cell nucleus into the cytoplasm where it will
be translated into the receptor protein.
2.38
T and B Cells
When stimulated by antigen, lymphocytes (see box on Cells of
the Immune System) first proliferate and then mature or differentiate
to become potent effector cells, such as B cells that release
antibodies or T cells that release cytokines. The normal T cell
proliferation that is seen when human lymphocytes and mouse splenocytes
(spleen cells) are exposed to antigens and mitogensj can be inhibited
by THC, 11-OH-THC, cannabinol, and 2-AG, as well as synthetic
cannabinoid agonists such as CP 55,940, WIN 55,212 and HU-210
61, 89, 93, 99, 127, 155 In contrast, one study testing anandamide
revealed little or no effect on T-cell proliferation.93
However, these drug effects are variable, and depend on experimental
conditions such as the experimental drug dose used, mitogen used,
percent of serum in the culture, and timing of cannabinoid drug
exposure. In general, lower doses of cannabinoids increase proliferation
while higher doses suppress proliferation. Doses that are effective
in suppressing immune function are typically greater than 10 µM
in cell culture studies and greater than 5 mg/kg in whole animal
studies.85 By comparison, at 0.05 mg/kg, people will experience
the full psychoactive effects of THC; however, because of their
high metabolic rates, small rodents frequently require drug doses
that are 100-fold higher than doses needed for humans to achieve
comparable drug effects. Thus, the immune effects of doses of
cannabinoids higher than those ever experienced by humans, should
be interpreted with caution.89, 93, 93, 127, 155
As with T cells, B cell proliferation can be suppressed by
various cannabinoids, such as THC, 11-OH-THC and 2-AG, but B cell
proliferation is more inhibited at lower drug concentrations than
T cell proliferation.89, 93 Conversely, low doses of THC, CP 55,940
and WIN 55,212-2 increase B-cell proliferation in cultured human
cells exposed to mitogen.35 This effect possibly involves the
CB2 receptor, because the effect appears to be the same when the
CB1 receptor was blocked by the antagonist, SR-141716A (which
does not block the CB2 receptor). The reason for the differences
in cell responsiveness to cannabinoids is probably due to differences
in cell type and source; for example, B cells collected from mouse
spleen might respond to cannabinoids somewhat differently than
B cells from human tonsils.
jMitogens are substances that stimulate cell division (mitosis)
and cell transformation.
2.39
Natural Killer Cells
Repeated injections of relatively low doses of THC (3 mg/kg/day
121 k) or two injections of a high dose (40 mg/kg86) suppress
the ability of natural killer (NK) cells to destroy foreign cells
in rats and mice. THC can also suppress natural killer cell cytolytic
activity in cell cultures; 11-OH-THC, is even more potent.86 In
contrast, THC doses below 10µM had no effect on natural killer
cell activity in mouse cell cultures.98
Macrophages
Macrophages perform various functions including phagocytosis
(ingestion and destruction of foreign substances), cytolysis,
antigen presentation to lymphocytes, and production of a variety
of active proteins involved in destroying microorganisms, tissue
repair and modulation of immune cells. Those functions can be
suppressed by THC doses similar to those capable of modulating
lymphocyte functions (see above).88, 109
Cytokines
Cytokines are proteins produced by immune cells. When released
from the producing cell they can alter the function of other cells
they come in contact with. In a sense, they are like hormones.
Thus, cannabinoids can either increase or decrease cytokine production
depending upon experimental conditions.
Certain cytokines, such as interferon- and interleukin-2 (IL-2)
are produced by T helper-1 (Th1) cells. These cytokines help to
activate cell-mediated immunity and the killer cells that eliminate
microbes from the body (see Box on cells of the immune system).
When injected into mice, THC suppresses the production of those
cytokines that modulate the host response to infection (see below).115
Cannabinoids also modulate interferons induced by viral infection,21
as well as other interferon inducers.85 Furthermore, in human
cell cultures, interferon production can be increased by low concentrations,
but decreased by high concentrations of either THC or cannabidiol.
6 In addition to Th1 cytokines, cannabinoids also modulate the
production of cytokines such as interleukin-1 (IL-1), tumor necrosis
factor (TNF), and interleukin-6 (IL-6). 145, 176 At 8 mg/kg, THC
can increase the in vivo mobilization of serum acute phase cytokines
including IL-1, TNF, and IL-6.90 Finally, although these studies
suggest that cannabinoids can induce an increase in cytokines,
other studies suggest that they can also suppress cytokine production.85
The different results might be due to different cell culture conditions
or because different cell lines were studied.
k While 3 mg/kg would be a high dose for humans (see table 3.1); in rodents, it is a low dose for immunological effects,
and a moderate dose for behavioral effects.
2.40
Antibody Production
Antibody production is an important measure of humoral immune
function as contrasted with cellular (cell-mediated immunity).
Antibody production can be suppressed in mice injected with relatively
low doses of THC (>5 mg/kg) or HU210 (>0.05 mg/kg) and in
mouse spleen cell cultures exposed to a variety of cannabinoids,
including THC, 11-OH-THC, cannabinol, cannabidiol, CP 55,940,
or HU-210.5, 6, 61, 78, 79, 84, 85, 142, 164 However, the inhibition of antibody response by cannabinoids was only observed when antibody-forming cells were exposed to T cell-dependent antigens (the responses require functional T cells and macrophages as accessory cells).
Conversely, antibody responses to several T cell-independent antigens
were not inhibited by THC, suggesting that the B cell is relatively
insensitive to inhibition by cannabinoids.142
Resistance To Infection In Animals Exposed To Cannabinoids
Bacterial infections evaluated in mice demonstrated that THC
can suppress resistance to infection, although the effect depends
upon the dose and timing of drug administration. Mice pre-treated
with THC (8 mg/kg) one day prior to infection with a sublethal
dose of the pneumonia-causing bacteria, Legionella pneumophilia,
and then treated again one day after the infection with THC, developed
symptoms of cytokine-mediated septic shock and died; control mice
that were not pre-treated with THC became immune to repeated infection
and survived the bacterial challenge.90 If only one injection
of THC was given or doses less than 5 mg/kg were used, all of
the mice survived the initial infection, but failed to survive
a subsequent challenge with a lethal dose of the bacteria; hence
these mice failed to develop immune memory in response to the
initial sublethal infection.87 Note that these are very high doses,
and are considerably higher than doses experienced by marijuana
users (see figure 3.1).115 In rats, doses of 4.0 mg/kg THC are
aversive95
Few studies have been done to evaluate the effect of THC on
viral infections, and this is an area that needs further study.20
Compared to healthy animals, THC might have greater immunosuppressive
effects in animals whose immune systems are severely weakened.
For example, a very high dose of THC (100 mg/kg) given twice,
two days before and after herpes simplex virus infection, was
shown to be a co-factor with herpes simplex virus in enhancing
the progression to death in an immunodeficient mouse model infected
with a leukemia virus.85 However, THC given as a single dose (100
mg/kg) two days before herpes simplex virus infection did not
promote the progression to death in these animals. Hence, whether
THC is immunosuppressive likely depends on the timing of THC exposure
relative to an infection.
2.41
Anti-inflammatory Effects
As discussed above, cannabinoid drugs can modulate the production of cytokines, which are central to inflammatory processes in the body. In addition, several studies have shown directly that cannabinoids can be anti-inflammatory. For example, in rats with autoimmune encephalomyelitis (an experimental model used to study multiple sclerosis), cannabinoids were shown to attenuate the signs and the symptoms of central nervous system damage.100, 172 (Some believe that nerve damage associated with multiple sclerosis is caused by an inflammatory reaction.) Likewise, the cannabinoid, HU-211, was shown to suppress brain inflammation that resulted from closed head injury 146 or infectious meningitis 7 in studies on rats. HU211 is a synthetic cannabinoid that does not bind to cannabinoid receptors, and is not psychoactive,7 thus, without direct evidence, the effects of marijuana cannot be assumed to include those of HU-211. CT-3, another atypical cannabinoid, suppresses acute and chronic joint inflammation in animals.178 It is a nonpsychoactive, synthetic derivative of 11-THC-oic acid (a breakdown product of THC), and does not appear to bind to cannabinoid receptors. 129 Cannabichromene, a cannabinoid found in marijuana, has also been reported to have anti-inflammatory properties.173 No mechanism of action for possible anti-inflammatory effects of cannabinoids has been identified and the effects of these atypical cannabinoids and effects of marijuana are not yet established.
It is interesting to note that two reports of cannabinoid-induced
analgesia are based on the ability of the endogenous cannabinoids,
anandamide and PEA, to reduce pain associated with local inflammation
that was experimentally induced by subcutaneous injections of
dilute formalin.22, 73 Both THC and anandamide can increase serum
levels of ACTH and corticosterone in animals.169 Those hormones
are involved in regulating many responses in the body, including
those to inflammation. The possible link between experimental
cannabinoid-induced analgesia and reported anti-inflammatory effects
of cannabinoids is important for potential therapeutic uses of
cannabinoid drugs, but has not yet been established.
Conclusions regarding effects on immune system
Based on cell culture and animal studies, cannabinoids have
been established as immunomodulators - that is, they increase
some immune responses and decrease others. The variable responses
depend upon experimental factors such as drug dose, timing of
delivery, and type of immune cell examined.
Cannabinoids affect multiple cellular targets within the immune
system and a variety of effector functions. Many of the effects
noted above appear to occur at
2.42
concentrations of > 5 µM in vitro and > 5 mg/kg in vivo.l
By comparison, a 5 mg injection of THC into a person (about 0.06
mg/kg) is enough to produce strong psychoactive effects. It should
be emphasized, however, that little is known about the effects
of chronic low dose exposure to cannabinoids on the immune system.
Another issue in need of further clarification involves the
potential usefulness of cannabinoids as therapeutic agents in
inflammatory diseases. Glucocorticoids have historically been
used for these diseases, but non-psychotropic cannabinoids potentially
have fewer side effects and might thus offer an improvement over
glucocorticoids in treating inflammatory diseases.
Conclusions and Recommendations
Following the progress of the past fifteen years in understanding
the effects of cannabinoids, research over the next decade is
likely to reveal even more. It is interesting to compare how little
we know about the cannabinoids with how much we know about the
opiates (table 2.8). This table, in fact, suggests good reason
for optimism about the future of cannabinoid drug development.
Now that many of the basic tools of cannabinoid pharmacology and
biology have been developed, one can expect to see rapid advances
that can begin to match what is known for opiate systems in the
brain.
Despite the tremendous progress in understanding the pharmacology
and neurobiology of brain cannabinoid systems, this field is still
in its early developmental stages. A key focus for future study
is the neurobiology of endogenous cannabinoids. Establishing the
precise brain localization - i.e.,., in which cells and where
in those cannabinoids are found, cellular storage and release
mechanisms, and uptake mechanisms will be crucial in determining
the biological role of this system. Technology that will be crucial
in establishing the biological significance of these systems will
be broad based and include such research tools as the transgenic,
or gene knockout mice, as have already been accomplished for various
opioid receptor types.26 In 1997, both CB1 and CB2 receptor knockout
mice were generated by a team of scientists at NIH, and a group
in France has developed another strain of CB1 in receptor knockout
mice.92
lIn vitro studies are those in which animal cells or tissue are
removed and studied outside the animal; in vivo studies are those
in which experiments are conducted in the whole animal.
2.43
Table 2.8 Historical comparisons between cannabinoids and opiates
| Comparisons between cannabinoids and opiates |
| . | Cannabinoids | Opiates |
Pharmacological
Discoveries |
| Discovery of receptor existence |
1988 (Howlett and Devane)36, 40 |
1973 (Pert and Snyder, Terenius, and Simon)123, 149, 162 |
| Identification of receptor antagonist |
1994 SR141716A (Rinaldi- Carmona)132 |
pre- 1973 Naloxone |
| Discovery of 1st endogenous ligand |
1992 Anandamide (Devane And Mechoulam)37 |
1975 Met- and Leu-enkephalin (Hughes et al)70 |
| 1st Receptor cloned | 1990 (Matsuda) 107 |
1992 (Evans et al. and Kieffer et al.)41, 82 |
| Natural functions of cannabinoid / opiate systems |
Unknown | Pain, reproduction, mood, movement, and others |
There are several research tools that will greatly aid such
investigations - in particular, a greater selection of agonists
and antagonists that permit discrimination between the activation
of CB1 versus CB2 receptors; hydrophilic agonists (that can be
delivered to animals or cells more effectively than hydrophobic
compounds). In the area of drug development, future progress should
continue to provide more specific agonists and antagonists for
CB1 and CB2 receptors, with varying potential for therapeutic
uses.
There are certain areas that will provide keys to a better
understanding of the potential therapeutic value of cannabinoids.
For example, basic biology indicates a role for cannabinoids in
pain and control of movement, which is consistent with a possible
therapeutic role in these areas. The evidence is relatively strong
for the treatment of pain, and intriguingly, although less well-established,
for movement disorders. The neuroprotective properties of cannabinoids
might prove therapeutically useful, although it should be noted
that this is a new area and other, better studied, neuroprotective
drugs have not yet been shown to be therapeutically useful. Cannabinoid
research is clearly relevant not only to drug abuse, but also
to
2.44
understanding basic human biology. Further, it offers the potential
for the discovery and development of new, therapeutically useful
drugs.
CONCLUSION: At this point, our knowledge about the biology
of marijuana and cannabinoids allows us to make some general conclusions:
Cannabinoids likely have a natural role in pain modulation,
control of movement, and memory.
The natural role of cannabinoids in immune systems is likely
multifaceted and remains unclear.
The brain develops tolerance to cannabinoids.
Animal research demonstrates the potential for dependence,
but this potential is observed under a narrower range of conditions
than with benzodiazepines, opiates cocaine, or nicotine.
Withdrawal symptoms can be observed in animals, but appear
to be mild compared to opiates or benzodiazepines, such as diazepam
(Valium ®).
CONCLUSION: The different cannabinoid receptor types found
in the body appear to play different roles in normal physiology.
In addition, some effects of cannabinoids appear to be independent
of those receptors. The variety of mechanisms through which cannabinoids
can influence human physiology underlies the variety of potential
therapeutic uses for drugs that might act selectively on different
cannabinoid systems.
RECOMMENDATION: Research should continue into the physiological
effects of synthetic and plant-derived cannabinoids and the natural
function of cannabinoids found in the body. Because different
cannabinoids appear to have different effects, cannabinoid research
should include, but not be restricted to effects attributable
to THC alone.
This chapter has summarized recent progress in understanding
the basic biology of cannabinoids, and provides a foundation for
the next two chapters which review studies on the potential health
risks (chapter 3) and benefits of marijuana use (chapter 4).
2.45
References
1. Abood ME, Martin BR. 1996. Molecular neurobiology of the
cannabinoid receptor. International Review of Neurobiology 39:197-221.
2. Abood ME, Sauss C, Fan F. Tilton CL, Martin BR. 1993. Development
of behavioral tolerance to delta 9- T HC without alteration of
cannabinoid receptor binding or mRNA levels in whole brain. Pharmacology,
Biochemistry and Behavior 46:575-9.
3. Aceto MD, Scates SM, Lowe JA, Martin BR. 1995. Cannabinoid
precipitated withdrawal by the selective cannabinoid receptor
antagonist, SR 141716A. European Journal of Pharmacology 282:R1-2.
4. Adams IB, Martin BR. 1996. Cannabis: pharmacology and toxicology in animals and humans. Addiction 91:1555-1614.
5. Baczynsky WO, Zimmerman AM. 1983a. Effects of delta-9-tetrahydrocannabinol, cannabinol, and cannabidiol on the immune system in mice: I. In
vivo investigation of the primary ant secondary immune response
Pharmocology 26: 1 - 11 .
6. Baczynsky WO, Zimmerman AM. 1983b. Effects of delta 9-tetrahydrocannabinol,
cannabinol and cannabidiol on the immune system in mice. II. In
vitro investigation using cultured mouse splenocytes. Pharmacology
26; 12-19.
7. Bass R. Engelhard D, Trembovler V, Shohami E. 1996. A novel
nonpsychotropic cannabinoid, HU-211, in the treatment of experimental
pneumococcal meningitis. Journal of Infectious Diseases 173:735-738.
8. Beardsley PM, Balster RL, Harris LS. 1986. Dependence on
tetrahydrocannabinol in rhesus monkeys. Journal of Pharmacology
and Experimental Therapeutics 239:311-319.
9. Ben-Shabat S. Fride E, Sheskin T. Tamiri T. Rhee MH, Vogel
Z. Bisogno T. De Petrocellis L, Di Marzo V, Mechoulam R. 1998.
An entourage effect: inactive endogenous fatty acid glycerol esters
enhance 2-arachidonoyl-glycerol cannabinoid activity. European
Journal of Pharmacology 353 :23-31.
10. Bennett GJ. 1994. Neuropathic Pain. In: Wall PD, Melzack
R Editors Textbook of Pain. Edinburgh: Churchill Livingstone.
11. Bird KD, Boleyn T. Chesher GB, Jackson DM, Starmer GA,
Teo RKC. 1980. Intercannabinoid and cannabinoid-ethanol interactions
and their effects on human performance. Psychopharmacology 71:181-188.
12. Bloom AS, Dewey WL, Harris LS, Brosius KK. 1977. 9-nor-9b-
hydroxyhexahydrocannabinol, a cannabinoid with potent antinociceptive
activity: Comparisons with morphine. Journal of Pharmacology and
Experimental Therapeutics 200:263-270.
13. Bornheim LM, Everhart ET, Li J. Correia MA. 1994. Induction
and genetic regulation of mouse hepatic cytochrome P450 by cannabidiol.
Biochemical Pharmacology (England) 48:161-171.
2.46
14. Bornheim LM, Kim KY, Chen B., Correia MA. 1993. The effect
of cannabidiol on mouse hepatic microsomal cytochrome P450-dependent
anandamide metabolism. Biochemical and Biophysical Research Communications
(United States) 197:740746.
15. Bornheim LM, Kim KY, Li J. Perotti BY, Benet LZ. 1995.
Effect of cannabidiol pretreatment on the kinetics of tetrahydrocannabinol
metabolites in mouse brain. Drug Metabolism and Disposition (United
States) 23:825-831.
16. Breivogel CS, Sim LJ, Childers SR. 1997. Regional differences
in cannabinoid receptor/G-protein coupling in rat brain. Journal
of Pharmacology and Experimental Therapeutics 282:1632-1642.
17. British Medical Association. 1997. Therapeutic uses of
cannabis. Amsterdam, The Netherlands: Harwood Academic Publishers.
18. Burkey THC Quock RM, Consroe P. Roeske WR, Yamamura H1.
1997. Delta-9 tetrahydrocannabinol is a partial agonist of cannabinoid
receptors in mouse brain. European Journal of Pharmacology 323:R3-R4.
19. Buxbaum DM. 1972. Analgesic activity of D9-tetrahydrocannabinol
in the rat and mouse. Psychopharmacology 25:275-280.
20. Cabral G A, Dove Pettit D A. 1998. Drugs and immunity:
cannabinoids and their role in decreased resistance to infectious
disease Journal of Neuroimmunology 83:116123.
21. Cabral GA, Lockmuller JC, Mishkin EM. 1986. Delta-9-tetrahydrocannabinol
decreases alpha/beta interferon response to herpes simplex virus
type 2 in the B6C3F1 mouse. Proceedings of the Society for Experimental
Biology and Medicine 181 :305-311.
22. Calignano A, La Rana G. Giuffrida A, Piomelli D. 1998.
Control of pain initiation by endogenous cannabinoids. Nature
394:277-281.
23. Campbell KA, Foster TC, Hampson RE, Deadwyler SA. 1986a.
Delta-9- tetrahydrocannabinol differentially affects sensory-evoked
potentials in the rat dentate gyrus. Journal of Pharmacology and
Experimental Therapeutics 239:936--940.
24. Campbell KA, Foster TC, Hapson RE, Deadwyler SA. 1986b.
Effects of delta-9- tetrahydrocannabinol on sensory-evoked discharges
of granule cells in the dentate gyrus of behaving rats. Journal
of Pharmacology and Experimental Therapeutics 239:941 -945.
25. Chen J. Marmur R. Pulles A, Paredes W. Gardner EL. 1993.
Ventral segmental microinjection of delta-9-tetrahydrocannabinol
enhances ventral segmental somatodendritic dopamine levels but
not forebrain dopamine levels: evidence for local neural action
by marijuana's psychoactive ingredient. Brain Research 621 :6570.
26. Childers SR. 1997. Opioid receptors: Pinning down the opiate
targets. Current Biology 7:R695-R697.
2.47
27. Childers SR, Breivogel CS. 1998. Cannabis and endogenous
cannabinoid systems. Drug and Alcohol Dependence 51:173-187.
28. Coffey RG, Yamamoto Y. Shella E, Pross S. 1996. tetrahydrocannabinol
inhibition of macrophage nitric oxide production. Biochemical
Pharmacology 52:743-751.
29. Collins DR, Pertwee RG, Davies SN. 1994. The action of
synthetic cannabinoids on the induction of long-term potentiation
in the rat hippocampal slice. European Journal of Pharmacology
259:R7-R8.
30. Collins DR, Pertwee RG, Davies SN. 1995. Prevention by
the cannabinoid antagonist, SR141716A, of cannabinoid-mediated
blockade of long-term potentiation in the rat hippocampal slice.
British Journal of Pharmacology 115:869-870.
31. Costa B. Parolaro D, Colleoni M. 1996. Chronic cannabinoid,
CP-55,940, administration alters biotransformation in the rat.
European Journal of Pharmacology 313: 17-24.
32. Daaka Y. Friedman H. Klein T. 1996. Cannabinoid receptor
proteins are increased in Jurkat, human T-Cell line after mitogen
activation. Journal of Pharmacology and Experimental Therapeutics
276:776-783.
33. Daaka Y. Zhu W. Friedman H. Klein T. 1997. Induction of
interleukin-2 receptor a gene by 9-tetrahydrocannabinol is mediated
by nuclear factor kB and CB1 cannabinoid receptor. DNA and Cell
Biology 16:301-309.
34. Deadwyler SA, Heyser CJ, Hampson RE. 1995. Complete adaptation
to the memory disruptive effects of delta-9-THC following 35 days
of exposure. Neuroscience Research Communications 17:9- 18.
35. Derocq JM, Segui M, Marchand J. Le Fur G. Casellas P. 1995.
Cannabinoids enhance human B-cell growth at low nanomolar concentrations.
FEBS Letters 369:177-182.
36. Devane WA, Dysarc FA, Johnson MR, Melvin LS, Howlett AC.
1988. Determination and characterization of a cannabinoid receptor
in rat brain. Molecular Pharmacology 34:605-613.
37. Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA,
Griffing F. Gibson D, Mandelbaum A, Etinger A, Mechoulam R. 1992.
Isolation and structure of a brain constituent that binds to the
cannabinoid receptor. Science 258: 1946- 1949.
38. Dewey WL. 1986. Cannabinoid pharamacology. Pharmacology
Review 38:151-178.
39. Di Chiara G. Imperato A. 1988. Drugs abused by humans preferentially
increase synaptic dopamine concentrations in the mesolimbic system
of freely moving rats. Proceedings of the National Academy of
Science, USA 85:5274-5278.
40. Dill JA, Howlett AC. 1988. Regulation of adenylate cyclase
by chronic exposure to cannabimimetic drugs. Journal of Pharmacology
and Experimental Therapeutics 244:1157-1163.
41. Evans DJ, Keith DEJ, Morrison H. Magendzo K, Edwards RH.
1992. Cloning of a delta
opioid receptor by functional expression. Science 258:1952- 1955.
2.48
42. Fan F. Tao Q. Abood ME, Martin BR. 1996. Cannabinoid receptor
down-regulation without alteration of the inhibitory effect of
CP 55,940 on adenylyl cyclase in the cerebellum of CP 55,940-tolerant
mice. Brain Research 706: 13-20.
43. Felder CC, Glass M. 1998. Cannabinoid receptors and their
endogenous agonists. Annual Reviews of Pharmacology and Toxicology
38: 179-200.
44. Felder CC, Nielsen A, Briley EM, Palkovits M, Priller J.
Axelrod J. Nguyen DN, Richardson JM, Riggin RM, Koppel GA, Paul
SM, Becker GW. 1996. Isolation and measurement of the endogenous
cannabinoid receptor agonist, anandamide, in brain and peripheral
tissues of human and rat. FEBS Letters 393:231 -5.
45. Fields HL. 1987. Pain. New York: McGraw-Hill.
46. Formukong EA, Evans AT, Evans FJ. 1988. Analgesic and anti-inflammatory
activity of constituents of Cannabis sativa L. Inflammation 12:361-371.
47. French ED. 1997. Delta-9-tetrabydrocannabinol excites rat
VTA dopamine neurons through activation of cannabinoid CB1 but
not opioid receptors. Neuroscience Letters 226:159-162.
48. Galiegue S. Mary S. Marchand J. Dussossoy D, Carriere D,
Carayon P. Bouaboula M, Shire D, Le Fur G. Casellas P. 1995. Expression
of central and peripheral cannabinoid receptors in human immune
tissues and leukocyte subpopulations. European Journal of Biochemistry
232:54-61
49. Gessa GL, Mascia MS, Casu MA, Carta G. 1997. Inhibition
of hippocampal acetylcholine release by cannabinoids: reversal
by SR 141716A. European Journal of Pharmacology 327:R1-2.
50. Gifford AN, Ashby Jr CR. 1996. Electrically evoked acetylcholine
release from hippocampal slices is inhibited by the cannabinoid
receptor agonist, WIN 55,212-2, and is potentiated by the cannabinoid
antagonist, SR 141716A. Journal of Pharmacology and Experimental
Therapeutics 277:1431-1436.
51. Gifford AN, Gardner EL, Ashby CRJ. 1997. The effect of
intravenous administration of delta-9-tetrahydrocannabinol on
the activity of A10 dopamine neurons recorded in vivo in anesthetized
rats. Neuropsychobiology 36:96-99.
52. Glass M, Dragunow M, Faull RLM. 1997. Cannabinoid receptors
in the human brain: a detailed anatomical and quantitative autoradiographic
study in the fetal, neonatal and adult human brain. Neuroscience
77:299-318.
53. Hadden JW, Hadden EM, Haddox MK, Goldberg ND. 1972. Guanosine
3':5'-cyclic monophosphates: A possible intracellular mediator
of mitogenic influences in Lymphocytes. Proceedings of the National
Academy of Sciences 69:3024-3027.
54. Hampson AJ, Grimaldi M, Axelrod J. Wink D. 1998. Cannabidiol
and (-)Delta-9- tetrahydrocannabinol are neuroprotective antioxidants.
Proceedings of the National Academy of Science of the United States
of America 95:8268-8273.
55. Haney M, Ward AS, Comer SD, Foltin RW, Fischman MW. 1999.
Abstinence symptoms following oral THC administration to humans.
Psychopharmacology 141:385-394.
2.49
56. Haney M, Ward AS, Comer SD, Foltin RW, Fischman MW. 1999.
Abstinence symptoms following smoked marijuana in humans. Psychopharmacology
141 :395-404.
57. Herkenham M. 1995. Localization of cannabinoid receptors
in brain and periphery. In: Pertwee RG ed. Cannabinoid Receptors.
Academic Press Limited. Pp. 145-166.
58. Herkenham M, Lynn AB, de Costa BR, Richfield EK. 1991a.
Neuronal localization of cannabinoid receptors in the basal ganglia
of the rat. Brain Research 547:267-274.
59. Herkenham M, Lynn AB, Johnson MR, Melvin LS, de Costa BR,
Rice KC. 1991b. Characterization and localization of cannabinoid
receptors in rat brain: a quantative in vitro autoradiographic
study. Journal of Neuroscience 11 :563-583.
60. Herkenham M, Lynn AB, Little MD, Johnson MR, Melvin LS,
de Costa BR, Rice KC. 1990. Cannabinoid receptor localization
in the brain. Proceedings of the National Academy of Sciences
of the United States of America 87: 1932- 1936.
61. Herring AC, Koh WS, Kaminski NE. 1998. Inhibition of the
cyclic AMP signaling cascade and nuclear factor binding to CRE
and kappa B elements by cannabinol, a minimally CNS-active cannabinoid.
Biochemical Pharmacology 55:1013-1023.
62. Henberg U. Eliav E, Bennett GJ, Kopin IJ. 1997. The analgesic
effects of R(+)-WIN 55,212-2 mesylate, a high affinity cannabinoid
agonist, in a rat model of neuropathic pain. Neuroscience Letters
221: 157-60.
63. Heyser CJ, Hampson RE, Deadwyler SA. 1993. Effects of delta-9-tetrahydrocannabinol on delayed match to sample performance in rats: Alterations in short-term memory associated with changes in task specific firing of hippocampal cells. Journal of Pharmacology and Experimental
Therapeutics 264:294-307.
64. Hohmann AG, Briley E M, Herkenham M. in press (1998). Pre-
and postsynaptic distribution of cannabinoid and mu opioid receptors
in rat spinal cord. Brain Research
65. Hohmann AG, Herkenham M. 1998. Regulation of cannabinoid
and mu opioid receptor binding sites following neonatal capsaicin
treatment. Neuroscience Letters 252: 1316.
66. Hohmann AG, Herkenham M. in press (1998). Localization
of central cannabinoid CB1 receptor mRNA in neuronal subpopulations
of rat dorsal root ganglia: A double- label in situ hybridization
study. Neuroscience
67. Hohmann AG, Martin WJ, Tsou K, Walker JM 1995 Inhibition
of noxious stimulus-evoked activity of spinal cord dorsal horn
neurons by the cannabinoid WIN 55,212-2. Life Sciences 56:2111-2119.
68. Hollister LE. 1986. Health aspects of cannabis. Pharmacological Reviews 38:1-20.
69. Hollister LE, Gillespie BA. 1975. Interactions in man of
delta-9-THC. II. Cannabinol and cannabidiol. Clinical Pharmacology
and Therapeutics 18:80-83.
2.50
70. Hughes J. Smith TW, Kosterlitz HW, Fothergill LA, Morgan
BA, Morris HR. 1975. Identification of two related pentapeptides
from the brain with potent opiate agonists activity. Nature 258:577-580.
71. Jackson AL, Murphy LL. 1997. Role of the hypothalamic-pituitary-adrenal
axis in the suppression of luteinizing hormone release by delta-9-tetrahydrocannabinol.
Neuroendocrinology 65:446-452.
72. Jacob J. Rarnabadran K, Campos-Medeiros M. 1981. A pharmacological
analysis of levonantradol antinociception in mice. Journal of
Clinical Pharmacology 21 :327S-333S.
73. Jaggar SI, Hasnie FS, Sellaturay S. Rice AS. 1998. The
anti-hyperalgesic actions of the cannabinoid anandamide and the
putative CB2 receptor agonist palmitoylethanolamide in visceral
and somatic inflammatory pain. Pain 76:189199.
74. Jeon YJ, Yang K-H, Pulaski JT, Kaminski NE. 1996. Attenuation
of inducible nitric oxide synthase gene expression by delta-9-tetrahydrocannabinol
is mediated through the inhibition of nuclear factor-kB/Rel activation.
Molecular Pharmacology 50:334341.
75. Johnson MR, Melvin LS. 1986. The discovery of non-classical
cannabinoid analgesics. In: Mechoulam R editor Cannabinoids as
therapeutic agents. Boca Raton, FL: CRC Press, Inc. Pp. 121-145.
76. Jones RT, Benowitz NL, Herning RI. 1981. Clinical relevance
of cannabis tolerance and dependence. Journal of Clinical Pharmacology
21: 143S-152S.
77. Kaminski NE. 1996. Immune regulation by cannabinoid compounds
through the inhibition of the cyclic AMP signaling cascade and
altered gene expression. Biochemical Pharmacology 52:1133-1140.
78. Kaminski NE, Abood M E, Kessler F K, Martin B R. Schatz
A R. 1992. ldentification of a functionally relevant cannabinoid
receptor on mouse spleen cells that is involved in cannabinoid-mediated
immune modulation. Molecular Pharmacology 42:736-742.
79. Kaminski NE, Koh WS, Yang KH, Lee M, Kessler FK. 1994 Suppression
of the humoral immune response by cannabinoids is partially mediated
through inhibition of adenylate cyclase by a pertussis toxin-sensitive
G-protein coupled mechanism. Biochemical Pharmacology 48:1899-1908.
80. Kaminski NE. 1998. Regulation of cAMP cascade, gene expression
and immune function by cannabinoid receptors. Journal of Neuroimmunology
83: 124- 132
81. Karniol IG, Shirakawa I, Kasinski N. Pfeferman A, Carlini
EA. 1975. Cannabidiol interferes with the effects of delta-9-tetrahydrocannbinol
in man. European Journal of Pharmacology 28:172-177.
82. Kieffer BL, Befort K, Gaveriaux-Ruff C, Hirth CG. 1992.
The delta-opioid receptor: isolation of a cDNA by expression clining
and pharmacological characterization. Proceedings of the National
Academy of Sciences of the United States of America 89: 12048-12052.
2.51
83. Kirby MT, Hampson RE, Deadwyler SA. 1995. Cannabinoids
selectively decrease paired pulse perforant path synaptic potentials
in the dentate gyrus in vitro. Brain Research 688:114-120.
84. Klein TW, Friedman H. 1990. Modulation of murine immune
cell function by marijuana components. In: Watson R ed. Drugs
of Abuse and immune Function. Boca Raton, FL: CRC Press.
85. Klein TW, Friedman H. Specter SC. 1998. Marijuana, immunity
and infection. Journal of Neuroimmunology 83: 102- 115.
86. Klein TW, Newton C, Friedman H. 1987. Inhibition of natural
killer cell function by marijuana components. Journal of Toxicology
and Environmental Health 20:321-332.
87. Klein TW, Newton C, Friedman H. 1994. Resistance to Legionella
pneumophila suppressed by the marijuana component, tetrahydrocannabinol
Journal of Infectious Diseases 169:1177-1179.
88. Klein TW, Newton C, Friedman H. 1998. Cannabinoid receptors
and immunity. Immunology Today 19:373-381.
89. Klein TW, Newton C, Widen R. Friedman H. l985 The effect
of delta-9 tetrahydrocannabinol and 11-hydroxy-delta-9-tetrahydrocannabinol
on T-lymphocyte and B-lymphocyte mitogen responses. Journal of
Immunopharmacology 7:451-466.
90. Klein TW, Newton C, Widen R. Friedman H. 1993. Delta-9-tetrahydrocannabinol
injection induces cytokine-mediated mortality of mice infected
with Legionella pneumophila. Journal of Pharmacology and Experimental
Therapeutics 267:635-640.
91. Koh WS, Yang KH, Kaminski NE. 1995. Cyclic AMP is an essential
factor in immune responses. Biochemical and Biophysical Research
Communications 206:703-709.
92. Ledent C, Valverde O. Cossu G. Petitet F. Aubert JF, Beslot
F in Bohme GA, Imperato A, Pedrazzini T. Roques BP, Vassart G.
Fratta W. Parmentier M. 1999. Unresponsiveness to cannabinoids
and reduced addictive effects of opiates in CB1 receptor knockout
mice. Science 283:401 -4 .
93. Lee M, Yang KH, Kaminski NE. 1995. Effects of putative
cannabinoid receptor ligands, anandamide and 2-arachidonyl-glycerol,
on immune function in B6C3F1 mouse splenocytes. Journal of Pharmacology
and Experimental Therupeutics 275:529536.
94. Lepore M, Liu X, Savage V, Matalon D, Gardner EL. 1996.
Genetic differences in delta 9- tetrahydrocannabinol-induced facilitation
of brain stimulation reward as measured by a rate-frequency curve-shift
electrical brain stimulation paradigm in three different rat strains.
Life Sciences 58:365-372
95. Lepore M, Vorel SR, Lowinson J. Gardner EL. 1995. Conditioned
place preference induced by delta 9-tetrahydrocannabinol: Comparison
with cocaine, morphine, and food reward. Life Sciences 56:2073-2080.
2.52
96. Lichtman AH, Martin BR. 1991a. Spinal and supraspinal
components of cannabinoid-induced antinociception. Journal of
Pharmacology and Experimental Therapeutics 258:517-523.
97. Little PJ, Compton DR, Mechoulam R. Martin BR. 1989. Stereochemical
effects of 11-OH- delta-8-THC-dimethylheptyl in mice and dogs.
Pharmacology Biochemistry Behavior 32:661-666.
98. Lu F. Ou DW. 1989. Cocaine or delta-9-tetrahydrocannabinol
does not affect cellular cytotoxicity in vitro. International
Journal of Pharmacology 11: 849-852.
99. Luo YD, Patel MK, Wiederhold MD, Ou DW. 1992. Effects of
cannabinoids and cocaine on the mitogen-induced transformations
of lymphocytes of human and mouse origins. International Journal
of lmmunopharmacology 14:49-56.
100. Lyman WD, Sonett JR, Brosnan CFER, Bornstein MB. 1989.
Delta 9-tetrahydrocannabinol: a novel treatment fur experimental
autoimmune encephalomyelitis. Journal of Neuroimmunology 23:73-81.
101. Mailleux P. Vanderhaeghen JJ. 1992. Distribution of neuronal
cannabinoid receptor in the adult rat brain: a comparative receptor
binding radioautography and in situ hybridization histochemistry.
Neuroscience 48:655-668.
102. Martin WJ, Hohmann AG, Walker JM. 1996. Suppression of
noxious stimulus-evoked activity in the ventral posterolateral
nucleus of the thalamus by a cannabinoid agonist: Correlation
between electrophysiological and antinociceptive effects. The
Journal of Neuroscience 16:6601 -6611.
103. Martin WJ, Patrick SL, Coffin PO, Tsou K, Walker JM. 1995.
An examination of the central sites of action of cannabinoid-induced
antinociception in the rat. Life Sciences 56:2103-2109.
104. Martin W J. Patrick S L, Coffin PO, Tsou K, Walker J M.
1995. An examination of the central sites of action of cannabinoid-induced
antinociception in the rat. Life Sciences 56:2103-2109.
105. Martin WJ, Tsou K, Walker JM. 1998. Cannabinoid receptor-mediated
inhibition of the rat tail-flick reflex after microinjections
into the rostral ventromedial medulla. Neuroscience Letters 242:33-36.
106. Martoletta MC, Cossu G. Fattore L, Gessa GL, Fratta W.
1998. Self-administration of the cannabinoid receptor agonist
WIN 55,212-2 in drug-naive mice. Neuroscience 85 :327-330.
107. Matsuda L, Lolait SJ, Brownstein MJ, Young AC, Bonner
TI. 1990. Structure of a cannabinoid receptor and functional
expression of the cloned cDNA. Nature 346:561 -564.
108. Mechoulam R. Ben-Shabat S. Hanus L, Ligumsky M, Kaminski
NSA, Gopher A, Almog S. Martin BR, Compton D, Pertwee RG, Griffin
G. Bayewitch M, Barg in Vogel Z. 1995. Identification of an endogenous
2-monoglyceride, present in canine gut, that binds to cannabinoid
receptors. Biochemical Pharmacology 50:83-90.
2.53
109. Mechoulam R. Hanus L, Fride E. 1998. Towards cannabinoid
drugs - revisited. In: Ellis GP, Luscombe DK, Oxford AW eds. Progress
in Medicinal Chemistry. v. 35. Amsterdam: Elsevier Science. Pp.
199-243.
110. Meng ID, Manning BH, Martin WJ, Fields HL. 1998. An analgesia
circuit activated by cannabinoids. Nature 395:381-383.
111. Miller AS, Walker JM. 1996. Electrophysiological effects
of a cannabinoid on neural activity in the globus pallidus. European
Journal of Pharmacology 304:29-35.
112. Munro S. Thomas KL, Abu-Shaar M. 1993. Molecular characterization
of a peripheral receptor for cannabinoids. Nature 365:61-65.
113. Murphy LL, Steger RW, Smith MS, Bartke A. 1990. Effects
of delta-9- tetrahydrocannabinol cannabinol and cannabidiol, alone
and in combinations, on luteinizing hormone and prolactin release
and on hypothalamic neurotransmitters in the male rat. Neuroendocrinology
52:316-321.
1l4. Narimatsu S. Watanabe K, Matsunaga T., Yamamoto I, Imaoka
S. Funae Y. Yoshimura H. 1993. Suppression of liver microsomal
drug-metabolizing enzyme activities in adult female rats pretreated
with cannabidiol Biological and Pharmaceutical Bulletin (Japan)
16:428-430.
115. Newton CA, Klein T. Friedman H. 1994. Secondary immunity
to Legionella pneumophila and Th1 activity are suppressed by delta-9-tetrabydrocannabinol
injection. Infection and immunity 62:4015-4020.
116. Norwicky AV, Teyler TJ, Vardaris RM. 1987. The modulation
of long-term potentiation by delta-9-tetrahydrocannabinol in the
rat hippocampus, in vitro. Brain Research Bulletin 19:663.
117. O'Leary D, Block RI, Flaum M, Boles Ponto LL, Watkins
GL, Hichwa RD. 1998. Acute marijuana effects on rCBF and cognition:
A PET study. Abstracts - Society for Neuroscience: 28th Annual
Meeting. Los Angeles, CA, November 7-12, l998.
Washington, DC: Society for Neuroscience.
118. Ohlsson A, Lindgren J-E, Wahlen A, Agurell S. Hollister
L E, Gillespie HK. 1980. Plasma delta-9-tetrahydrocannabinol concentrations
and clinical effects after oral and intravenous administration
and smoking. Clinical Pharmacology and Therapeutics
28:409-416.
119. Oviedo A, Glowa J. Herkenham M. 1993. Chronic cannabinoid
administration alters cannabinoid receptor binding in rat brain:
a quantitative autoradiographic study. Brain Research 616:293-302
120. Pacheco MA, Ward SJ, Childers SR. 1993. Identification
of cannabinoid receptors in cultures of rat cerebellar granule
cells. Brain Research 603: 102- 110.
121. Patel V, Borysenko M, Kumar MSA, Millard WJ. 1985. Effects
of acute and subchronic delta-9-tetrahydrocannabinol administration
on the plasma catecholamine, beta- endorphin, and corticosterone
levels and splenic natural killer cell activity in rats.
Proceedings of the Society for Experimental Biology and Medicine
180:400-404.
2.54
122. Pepe S. Ruggiero A, Tortora G. Ciaardiello F. Garbi C,
Yokozaki H. Cho-Chung YS, Clair T. Skalhegg BS, Bianco AR. 1994.
Flow cytometric detection of the RT alpha subunit of type-I cAMP-dependent
protein kinase in human cells. Cytometry 15:7379.
123. Pert CB, Snyder SH. 1973. Opiate receptor: demonstration
in nervous tissue. Science 179:1011-1014.
124. Pertwee RG. 1997b. Pharmacology of cannabinoid CB1 and
CB2 receptors. Pharmacology and Therapeutics 74: 129-180.
125. Pertwee RG, Stevenson LA, Griffin G. 1993. Cross-tolerance
between delta-9- tetrahydrocannabinol and the cannabimimetic agents,
CP 55,940, WIN 55,212-2 and anandamide [published erratum appears
in British Journal of Pharmacology 1994 Mar; 111(3):968]. British
Journal of Pharmacology l10:1483-90.
126. Pertwee RG, Wickens AP. 1991. Enhancement by chlordiazepoxide
of catalepsy induced in rats by intravenous or intrapallidal injections
of enantiomeric cannabinoids. Neuropharmacology 30:237-244.
127. Pross SH, Nakano Y. Widen R. McHugh S. Newton C, Klein
TW, Friedman H. 1992. Differing effects of delta-9-tetrahydrocannabinol
(THC) on murine spleen cell populations dependent upon stimulators
International Journal of Immunopharmacology 14:1019- 1027.
128. Razdan RK. 1986. Structure-activity relationships in cannabinoids.
Pharmacology Review 38 75-149.
129. Rhee MH, Vogel Z. Barg J. Bayewitch M, Levy R. Hanus L,
Breuer A, Mechoulam R. 1997. Cannabinol derivatives: binding to
cannabinoid receptors and inhibition of adenylcyclase. Journal
of Medicinal Chemistry 40:3228-3233.
130. Richardson JD, Aanonsen L, Hargreaves KM. 1998. Hypoactivity
of the spinal cannabinoid system results in NMDA-dependent hyperalgesia.
Journal of Neuroscience 18:451--457.
131. Richardson JD, Kilo S. Hargreaves KM. 1998. Cannabinoids
reduce hyperalgesia and inflammation via interaction with peripheral
CB1 receptors. Pain 75:111 - 119.
132. Rinaldi-Carmona M, Barth F. Heaulme M, Shire D, Calandra
B. Congy C, Martinez S. Maruani J. Neliat G. Caput D, Ferrara
P. Soubrie P. Breliere JC, Le Fur G. 1994. SR141716A, a potent
and selective antagonist of the brain cannabinoid receptor. FEBS
Letters 350:240-244.
133. Rinaldi-Carmona M, Barth F. Millan J. Defrocq J. Casellas
P. Congy C, Oustric D, Sarran M, Bouaboula M, Calandra B. Portier
M, Shire D, Breliere J. Le Fur G. 1998. SR144528, the first potent
and selective antagonist of the CB2 cannabinoid receptor. Journal
of Pharmacology and Experimental Therapeutics 284:644-650.
134. Rodriguez de Fonseca F. Fernandez-Ruiz JJ, Murphy LL,
Eldridge JC, Steger RW, Bartke A. 1991. Effects of delta-9-tetrahydrocannabinol
exposure on adrenal medullary function: evidence of an acute effect
and development of tolerance in chronic treatments. Pharmacology,
Biochemistry and Behavior 40:593-598
2.55
135. Rodriguez de Fonseca F. Carrera MRA, Navarro M, Koob
G. Weiss F. 1997. Activation of corticotropin-releasing factor
in the limbic system during cannabinoid withdrawal [see comments
Science 1997. 276:1967-1968]. Science 276:2050-2054.
136. Rodriguez de Fonseca F. Gorriti MA, Fernandez-Ruiz JJ,
Palomo T. Ramos JA. 1994. Down-regulation of rat brain cannabinoid
binding sites after chronic delta-9- tetrahydrocannabinol treatment.
Pharmacology, Biochemistry and Behavior 47:33--40.
137. Romero J. Garcia L, Fernandez-Ruiz JJ, Cebeira M, Ramos
JA 1995 Changes in rat brain cannabinoid binding sites after acute
or chronic exposure to their endogenous agonist, anandamide, or
to delta-9-tetrahydrocannabinol. Pharmacology, Biochemistry and
Behavior 51:731 -737.
138. Romero J. Garcia-Palomero E, Castro JG, Garcia-Gil L,
Ramos JA, Fernandez-Ruiz JJ. 1997. Effects of chronic exposure
to delta-9-tetrahydrocannabinol on cannabinoid receptor binding
and mRNA levels in several rat brain regions. Molecular Brain
Research 46: 100-8.
139. Russell DH. 1978. Type I cyclic AMP-dependent protein
kinase as a positive effector of growth. Advances in Cyclic Nucleotide
Research 9:493-506.
140. Sanudo-Pena M C, Tsou K, Delay E R. Hohmann AG, Force
M, Walker JM. 1997. Endogenous cannabinoids as an aversive or
counter-rewarding system in the rat. Neuroscience Letters 223:
125- 128.
141. Sanudo-Pena MC, Walker JM. 1997. Role of the subthalamic
nucleus in cannabinoid actions in the substantia nigra of the
rat. Journal of Neurophysiology 77: 1635-1638.
142. Schatz AR, Koh WS, Kaminski, NE. 1993. Delta-9-tetrahydrocannabinol
selectively inhibits T-cell dependent humoral immune responses
through direct inhibition of accessory T-cell function. Immunopharmacology
26: 129- 137.
143. Schlicker E, Timm J. Zenter J. Goethert M. 1997. Cannabinoid
CB1 receptor-mediated inhibition of noradrenaline release in the
human and guinea-pig hippocampus. Naunyn-Schmiedeberg's Archives
of Pharmacology 356:583-589.
144. Shen M, Piser TM, Seybold VS, Thayer SA. 1996. Cannabinoid
receptor agonists inhibit glutamatergic synaptic transmission
in rat hippocampal cultures. Journal of Neuroscience 16:4322-4334.
145. Shivers S C, Newton C, Friedman H. Klein TW. 1994. Delta
9-tetrahydrocannabinol (THC) modulates IL-1 bioactivity in human
monocyte/macrophage cell lines. Life Sciences 54: 1281-1289.
146. Shohami E, Gallily R. Mechoulam R. Bass R. Ben-Hur T.
1997. Cytokine production in the brain following closed head injury:
dexanabinol (HU-211) is a novel TNF-alpha inhibitor and an effective
neuroprotectant. Journal of Neuroimmunology 72: 169--177.
147. Sim LJ, Hampson RE, Deadwyler SA, Childers SR. 1996. Effects
of chronic treatment with delta-9-tetrahydrocannabinol on cannabinoid-stimulated
[35S]GTPyS autoradiography in rat brain. Journal of Neuroscience
16:8057-8066.
2.56
148. Sim LJ, Xiao R. Selley DE, Childers SR. 1996. Differences
in G-protein activation by mu- and delta-opioid, and cannabinoid,
receptors in rat striatum. European Journal of Pharmacology 307:97-105.
149. Simon EJ. 1973. In search of the opiate receptor. American
Journal of Medical Sciences 266: 160- 168.
150. Skaper SD, Buriani A, Dal Toso R. Petrelli L, Romanello
S. Facci L, Leon A. 1996. The ALIAmide palmitoylethanolamide and
cannabinoids, but not anandamide, are protective in a delayed
postglutamate paradigm of excitotoxic death in cerebellar granule
neurons. Proceedings of the National Academy of Sciences of the
United States of America 93:3984-9.
151. Smith JW, Steiner AL, Newberry WM, Parker CW. 1971. Cyclic
adenosine 3',5'- monophosphate in human lymphocytes: Alteration
after phytohemagglutinin. Journal of Clinical Investigation 50:432-441.
152. Smith PB, Compton DR, Welch SP, Razdan RK, Mechoulam R.
Martin BR. 1994. The pharmacological activity of anandamide, a
putative endogenous cannabinoid, in mice. Journal of Pharmacology
and Experimental Therapeutics 270:219-227.
153. Smith PB, Welch SP, Martin BR. 1994. Interactions between
delta 9-tetrahydrocannabinol and kappa opioids in mice. Journal
of Pharmacology and Experimental Therapeutics 268:1381-1387.
154. Sofia RD, Nalepa SD, Harakal JJ, Vassar HB. 1973. Anti-edema
and analgesic properties of delta-9-tetrahydrocannabinol (THC).
Journal of Pharmacology and Experimental Therapeutics 186:646-655.
155. Specter S. Lancz C, Hazelden J. 1990. Marijuana and immunity:
tetrahydrocannabinol mediated inhibition of lymphocyte blastogenesis.
International Journal of Immunopharmacology 12;261-267.
156. Stefano G. Salzet B. Salzet M. 1997. Identification and
characterization of the leech CNS cannabinoid receptor: Coupling
to nitric oxide release. Brain Research 753:219--224.
157. Stella N. Schweitzer P. Piomelli D. 1997. A second endogenous
cannabinoid that modulates long term potentiation. Nature 388:773-778.
158. Strangman NM, Patrick SL, Hohmann AG, Tsou K, Walker JM.
1998. Evidence for a role of endogenous cannabinoids in the modulation
of acute and tonic pain sensitivity. Brain Research 813:. 323-328.
159. Sulcova C, Mechoulam R., Fride E. 1998. Biphasic effects
of anandamide. Pharmacology, Biochemistry and Behavior 59:347-352.
160. Szabo B. Dorner L, Pfreundtner C, Norenberg W. Starke
K. 1998. Inhibition of GABAergic inhibitory postsynaptic currents
by cannabinoids in rat corpus straitum Neuroscience 85:395-403.
161. Tanda G. Pontieri FE, Di Chiara G. 1997. Cannabinoid and
heroin activation of mesolimbic dopamine transmission by a common
µ1 opioid receptor mechanism. Science 276:2048-2049.
2.57
162. Terenius L. 1973. Characteristics of the "receptor"
for narcotic analgesics in synaptic plasma membrane fraction from
rat brain. Acta Pharmacologica Et Toxicologica 33:377-384.
163. Terranova JP, Michaud JC, Le Fur G. Soubrie P. 1995. Inhibition of long-term potentiation in rat hippocampal slice by anandamide
and WIN 55.212-2: reversal by SR141716 A, a selective antagonist
of CB1 cannabinoid receptors. Naunyn-Schmiedeberg's Archives of Pharmacology 352:576-579.
164. Titishov N. Mechoulam R. Zimmerman AM. 1989. Stereospecific
effects of (-) and (+)-7- hydroxy-delta-6-tetrahydrocannabinol-dimethylheptyl
on the immune system of mice. Pharmacology 39:337-349.
165. Tsou K, Brown S. Sanudo-Pena MC, Mackie K, Walker JM.
1998. Immunohistochemical distribution of cannabinoid CB1 receptors
in the rat central nervous system. Neuroscience 83:393-411.
166. Tsou K, Patrick SL, Walker JM. 1995. Physical withdrawal
in rats tolerant to delta-9- tetrahydrocannabinol precipitated
by a cannabinoid receptor antagonist. European Journal of Pharmacology
280:R13-R15.
167. Watson PF, Krupinski J. Kempinski A, Frankenfield C. 1994.
Molecular cloning and characterization of the type VII isoform
of mammalian adenylyl cyclase expressed widely in mouse tissues
and in S49 mouse Iymphoma cells. Journal of Biological Chemistry
269:28893-28898.
168. Watzl B. Scuder P. Watson RR. 1991. Marijuana components
stimulate human peripheral blood mononuclear cell secretion of
interferon-gamma and suppress interleukin-1 alpha in vitro. International
Journal of Immunopharmology 13:1091-1097.
169. Weidenfeld J. Feldman S. Mechoulam R. 1994. Effect of
the brain constituent anandamide, a cannabinoid receptor agonist,
on the hypothalamo-pituitary-adrenal axis in the rat. Neuroendocrinology
59:110-112.
170. Welch SP. 1993. Blockade of cannabinoid-induced antinociception
by norbinaltorphimine, but not N,N-diallyl-tyrosine-Aib-phenylalanine-leucine,
ICI 174,864 or naloxone in mice. Journal of Pharmacology and Experimental
Therapeutics 265:633-640.
171. Welch SP, Thomas C, Patrick GS. 1995. Modulation of cannabinoid-induced
antinociception after intracerebroventricular versus intrathecal
administration to mice: Possible mechanisms for interaction with
morphine. Journal of Clinical and Experimental Therapeutics 272:310-321.
172. Wirguin I, Mechoulam R. Breuer A, Schezen E, Weidenfeld
J. Brenner T. 1994. Suppression of experimental autoimmune encephalomyelitis
by cannabinoids. Immunopharmacology 28 :209-214.
173. Wirth PW, Watson ES, ElSohly M, Turner CE, Murphy JC.
1980. Anti-inflammatory properties of cannabichromene. Life Sciences
26:1991-1995.
174. Yaksh TL. 1981. The antinociceptive effects of intrathecally
administered levonantradol and desacetyl-levonantradol in the
rat. Journal of Clinical Pharmacology 21 :334S--340S.
2.58
175. Yoshida H. Usami N. Ohishi Y. Watanabe K, Yamamoto I,
Yoshimura H. 1995. Synthesis and pharmacological effects in mice
of halogenated cannabinol derivatives. Chemical and Pharmaceutical
Bulletin 42:335-337.
176. Zhu W. Newton C, Daaka Y. Friedman H. Klein TW. 1994.
Delta 9-tetrahydrocannabinol enhances the secretion of interleukin
I from endotoxin-stimulated macrophages. Journal of Pharmacology
and Experimental Therapeutics 270: 1334-1339.
177. Zuardi AW, Shirakawa I, Finkelfarb E, Karniol IG. 1982.
Action of cannabidiol on the anxiety and other effects produced
by delta 9-THC in normal subjects. Psychopharmacology (Berl) 76:245-250.
178. Zurier R.B., Rossetti RG, Lane JH, Goldberg JM, Hunter
SA, Burstein SH 1998. Dimethylheptyl-THC- 11 oic acid: A non-psychoactive
antiinflmnmatory agent with a cannabinoid template structure.
Arthritis and Rheumatism 41:163- 170.
2.59
Chapter 3 ==>>